circRNA 最初研究的很少,只有很小一部分基因有检测到circRNA, 当时都认为是剪切错误形成的,对于其功能也没人去研究;学者对人类的成纤维细胞进行转录组测序,构建去核糖体文库, 同时采用了RNase酶消化线性RNA 和 不消化线性RNA的两种文库,同时检测circRNA , 最终检测到了25000 多种circRNA ,这些circRNA 来源于exon 区, 叫做 ecircRNA, 保守估计,有14%的基因都产生了circRNA. 利用同样的方法,在小鼠睾丸组织中,鉴定到了69种和人类的circRNA 高度同源的circRNA。circRNA 的表达量,可以被siRNA 调控,推测具有竞争性内源RNA的作用,而且通过生物信息学手段发现,这些ecircRNA 大部分都具有 ALU 重复元件,这些都表明circRNA 并不是随机的剪切错误产生的,而是固有的一类,种类丰富,结构稳定,序列保守的内源性RNA;
真核生物的total RNA 中,非编码RNA(ncRNA) 占据了95%的比例,尽管在ncRNA 中,rRNA 和 tRNA 占据了绝大部分,但是其他类型的ncRNA , 比如miRNA和 lncRNA , 其研究越来越多,越来越受到重视,科学家通过RNase R 消化线性RNA, 利用高通量测序来研究circRNA, 主要使用了mapsplice 工具。
为了研究哺乳动物细胞中的circRNA , 科学家发明了一种 CircleSeq 的文库构建方法
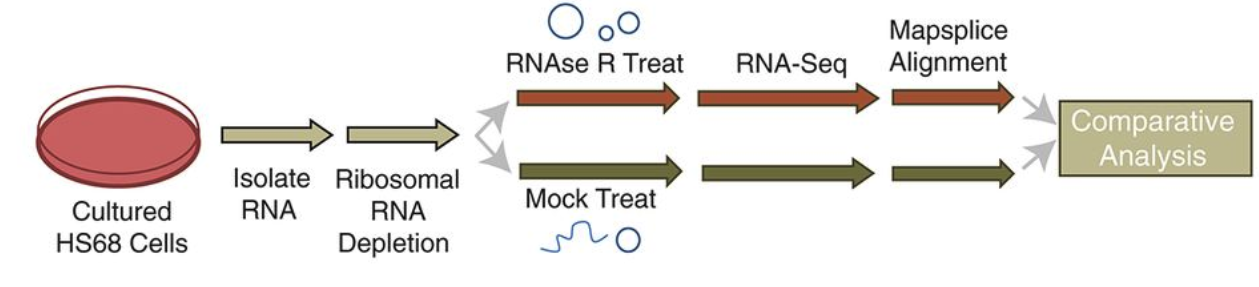
首先去除rRNA, 然后用RNAse R 消化线性RNA, 同时还设立了对照组,就是不消化线性RNA的文库作为对照;
研究发现 RNase R 处理的文库,可以很好的实现circRNA 富集的效果,更加有利于检测 低风度的circRNA , 富集效果是普通文库的10倍以上;
Hiseq 对每个样本进行高通量测序,数据量为300M,使用mapsplice 软件比对参考基因组,
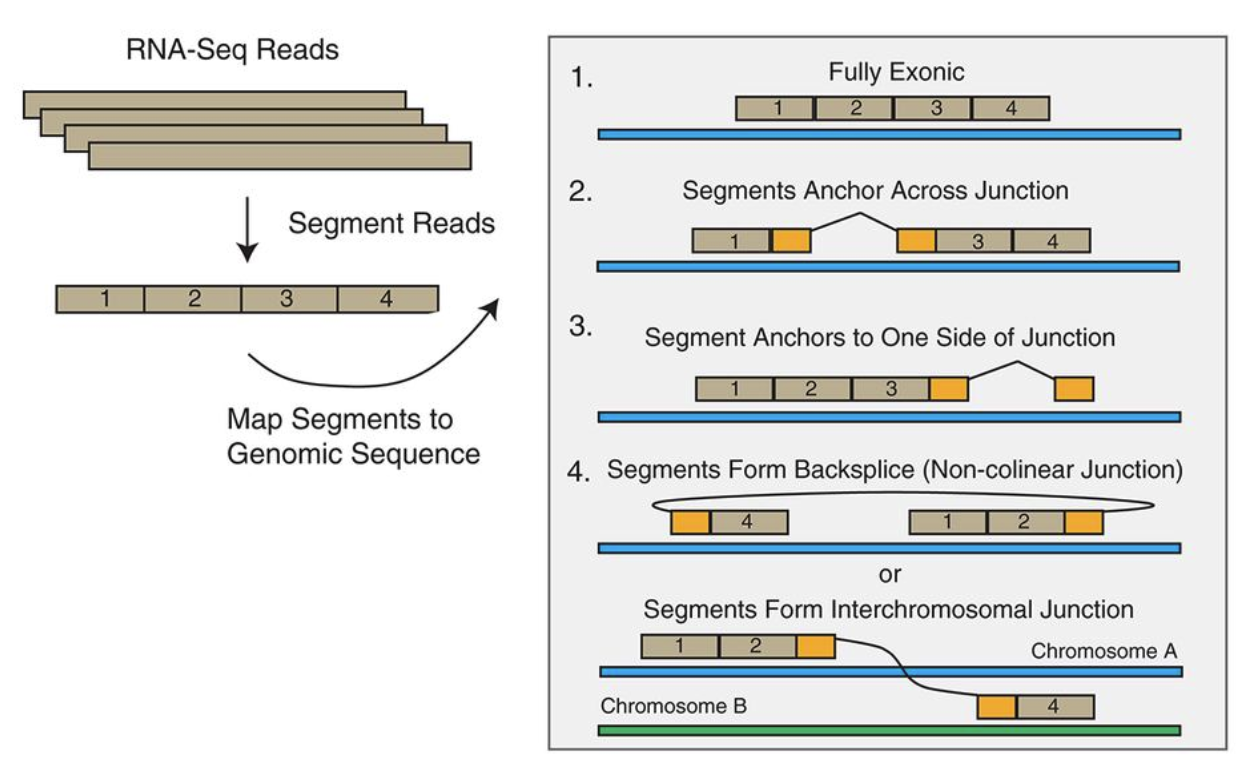
对于每一条测序的reads, mapsplice 会一次进行下列 4种比对:
1) 完全来自1个exon , 这样的reads 直接比对上参考基因组
2)跨越了剪切位点,叫做junction reads, 这样的reads 在剪切位点的两侧,分别能够比对上参考基因组;
3)反向剪切 backsplice 生成的reads, 同样是在剪切位点两侧分别比对,但是比对的顺序和线性RNA的顺序正好相反;
4)融合转录本的序列,融合位点两侧的序列比对上了不同的染色体
参考资料:
http://rnajournal.cshlp.org/content/19/2/141.long#F6