原文链接:http://tecdat.cn/?p=27258
原文出处:拓端数据部落公众号
相关视频:马尔可夫链原理可视化解释与R语言区制转换Markov regime switching实例
马尔可夫链原理可视化解释与R语言区制转换Markov regime switching实例
,时长07:25
相关视频
马尔可夫链蒙特卡罗方法MCMC原理与R语言实现
,时长08:47
摘要
本文开发和应用用于生物序列分析的隐马尔可夫模型和HMM。它包含多个和成对序列比对、模型构建和参数优化、文件导入/导出、实现条件序列概率的前向、后向和 Viterbi 算法、基于树的序列加权和序列模拟的功能。
介绍
隐马尔可夫模型 (HMM) 是计算生物学中许多最重要任务的基础,包括多序列比对、基因组注释以及越来越多的序列数据库搜索。最初是为语音识别算法开发的,由于计算能力的进步使得完全概率分析代替启发式近似成为可能,因此它们在分子生物学领域的应用急剧增加。
在这里,我们展示了 用于在 R 环境中分析隐藏马尔可夫模型。
隐马尔可夫模型
隐马尔可夫模型是一个序列或一组序列的概率数据生成机制。它由 状态网络描绘,其值特定于每个状态。这些状态由一组互连的 转换概率遍历,其中包括保持在任何给定状态的概率以及转换到每个其他连接状态的概率。
Durbin et al (1998) 第 3.2 章给出了一个简单 HMM 的例子。一个假想的赌场有两个骰子,一个公平的,一个加权的。公平骰子从字母表 {1, 2, 3, 4, 5, 6} 中以相等的概率发出(每个 1/6)。加权骰子掷出“6”的概率为 0.5,而其他 5 个的概率为 0.1。如果庄家有公平骰子,他可能会在每次掷骰后以 0.05 的概率偷偷切换到装好的骰子,留下 95% 的机会保留公平骰子。或者,如果他有加权的骰子,他将以 0.1 的概率切换回公平骰子,或者更有可能以 0.9 的概率保留加权骰子。
这个例子可以用一个简单的两态隐马尔可夫模型来表示。以下代码手动构建和绘制“HMM”对象。
-
-
### 定义转移概率矩阵A
-
diamnsames(A) <- lidst(from = sdtatases, to = statddes)
-
### 定义概率矩阵 E
-
dimnsdamesas(E) <- aslist(sdtateass = stasdates[-1])
-
### 创建 HMM 对象
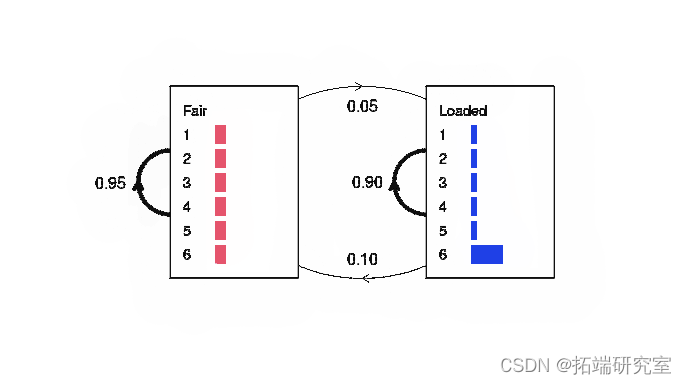 图 1:不诚实赌场示例的简单隐马尔可夫模型。 plot.HMM 方法将转换概率描述为加权线。在这个例子中没有模拟开始/结束状态。
图 1:不诚实赌场示例的简单隐马尔可夫模型。 plot.HMM 方法将转换概率描述为加权线。在这个例子中没有模拟开始/结束状态。
对于一系列观察到的掷骰,我们可以使用维特比算法建立最可能的隐藏状态序列(包括最有可能发生骰子切换的时间)。在 Durbin 等人 (1998) 第 3.2 章给出的示例中,观察到的 300 次滚动的序列为:



一些可观察到的 6 簇表明加载的骰子在某个阶段出现了,但是骰子切换是什么时候发生的呢?在下面的代码中,Viterbi 算法用于在给定模型的情况下找到最可能的隐藏状态序列。
-
c("Fd", "La")[mastch(namdes(casasinao), c("Fsair", "Loasadded"))]
-
### 找到预测路径
-
viat1 <- Vitedsbi(xasd)
-
预测 <- c("Fa", "aL")[vist1$dpaath + 1]
-
### 注意输出维特比对象的路径元素是一个整数向量
将预测路径与实际隐藏序列进行比较,Viterbi 算法并不遥远:

我们还可以使用 forward 和/或 backward 算法计算给定模型的序列的完整概率和后验概率:
-
casasino.dspost <asd- posterassdior(xa, asdcasino)
-
plot(1:300, saseq(0, 1, lasdength.ouast = 300)
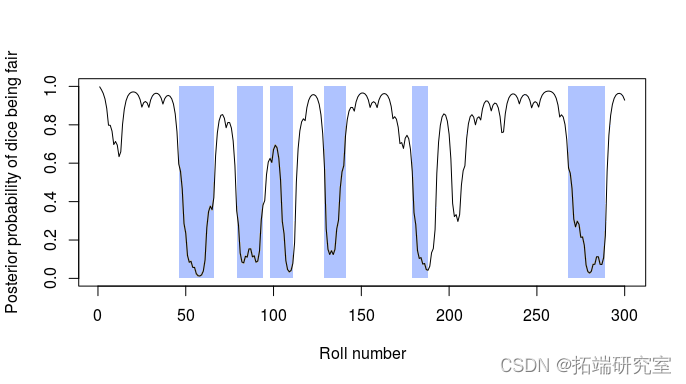 图 2:300 次掷骰子的后验状态概率。 这条线显示了骰子在每次掷骰时都是公平的后验概率,而灰色矩形显示了使用加载骰子的实际周期。有关详细信息,请参见 Durbin 等人 (1998) 第 3.2 章。
图 2:300 次掷骰子的后验状态概率。 这条线显示了骰子在每次掷骰时都是公平的后验概率,而灰色矩形显示了使用加载骰子的实际周期。有关详细信息,请参见 Durbin 等人 (1998) 第 3.2 章。
从序列数据中导出 HMM
从一组训练序列构建HMM 。以下代码从我们的单个掷骰子序列及其已知状态路径(存储为序列的“名称”属性)中派生出一个简单的 HMM。
-
yajd <- dergsiveHMhM(lissffgtd(cadsfihnos), logshasdpfgace = FALSE)
-
plosgt(y, teagghxteasfdxp = 1.5)
-
-
### 可选择将转换概率添加为文本
-
tedhsaxt(xda = 0.02, yad = 0.5, ladbelass = roasdund(ya$Aa["Fasir", "Fadir"], 2))
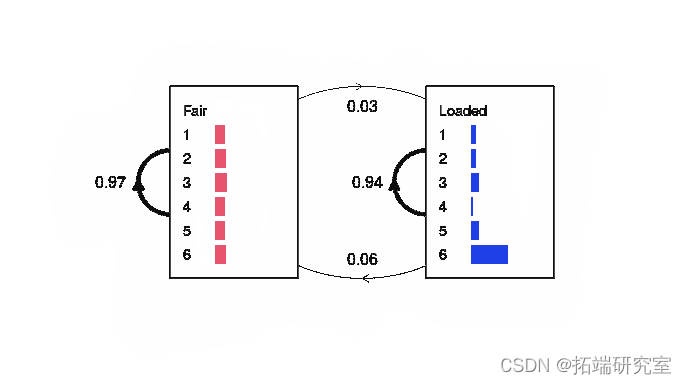
图 3:从 300 次掷骰子序列派生的简单 HMM。 如图 所示,转换概率显示为加权线,发射概率显示为水平灰色条。
尽管训练数据仅由一个序列组成,但这似乎与实际模型相当接近。人们通常会从许多此类序列的列表中导出 HMM(因此输入参数是列表而不是向量的原因),但为清楚起见,此示例已简化。
HMM隐马尔可夫模型
Profile HMM马尔可夫模型是标准 HMM 的扩展,其中转移概率是 特定于位置的。也就是说,它们可以在序列中的每个点发生变化。这些模型通常比其简单的 HMM 模型具有更多的参数,但对于序列分析可能非常强大。Profile HMM 的前身通常是多序列比对。
图 4 将上面列出的三种状态类型分别显示为圆形、菱形和矩形。这些状态由图中的加权线所示的转移概率链接。
考虑来自 Durbin 等人 (1998) 第 5.3 章的氨基酸序列的小部分比对:
-
data(gloasdbins)
-
gldobinasds
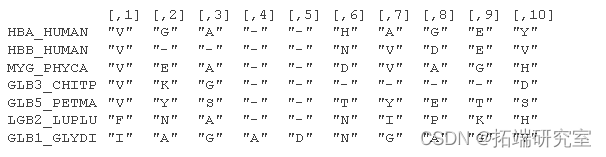
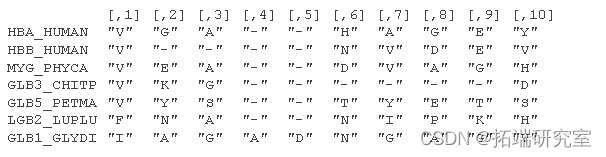
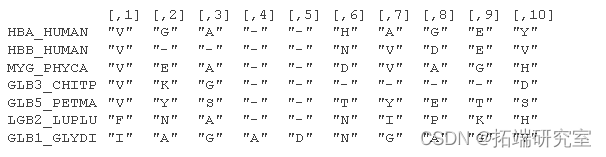
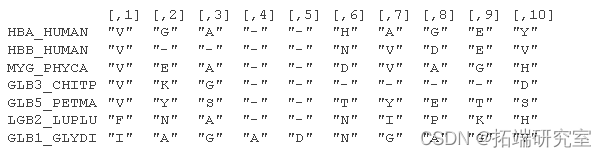
位置特定模式包括在位置 1 观察到“V”和在位置 3 观察到“A”或“G”的高概率。
以下代码从 globin 数据派生出 HMM 轮廓并绘制模型:
glsosdbgnss.PsdHMMf <- divfgsesPHMM(glns, resiufes = "AMaIsdfNO", pdfntss = "Laplsaace")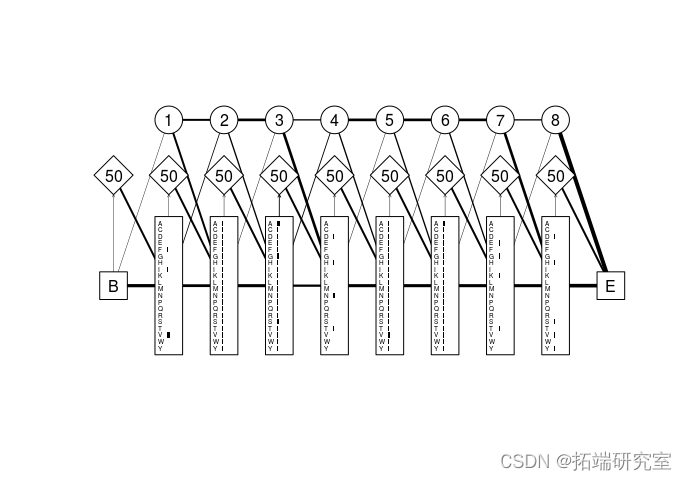
图 4:源自部分珠蛋白序列比对的轮廓 HMM。
匹配状态显示为矩形,插入状态显示为菱形,删除状态显示为圆形。灰色水平条表示模型中每个位置的字母表(在本例中为氨基酸字母表)中每个残基的发射概率。删除状态中的数字只是模型模块编号,而插入状态中的数字是在下一个发射周期保持在当前插入状态的概率。在必要时对线进行加权和定向,以反映状态之间的转换概率。大的“B”和“E”标签分别代表静默开始和结束状态。
我们可以通过计算该序列在模型中的最优路径来证明这一点,同样使用维特比算法:
-
padaath <- Vitferbi(glsdosbinsd.PsHMM, glodbdins["GsLB1d_GLYsDI", ])$patdh
-
pdatsh
![]()
Viterbi 对象的“路径”元素是一个整数向量,其元素取值为 0(“删除”)、1(“匹配”)或 2(“插入”)。路径可以更直观地表示为字符而不是索引,如下所示:
c("D", "M", "I")[pgatsh + 1]![]()
请注意,向每个路径元素添加 1 只是为了将 C/C++ 索引样式(从 0 开始)转换为 R 的样式。
序列模拟
为了模拟随机变化的数据。例如,以下代码模拟了来自小珠蛋白 HMM 的 10 个随机序列的列表:
-
siasdm <- ldist(lfenhgsth = 10)
-
suppressWarffnings(RNGvghjerhhjion("3.5.0"))
-
k
-
for(i in 1:10) shim[[i]] <- genkerate(globihhgkjknks.PHhMM, size = 20)
-
sihmg


模型训练
使用 Baum Welch 或 Viterbi 训练算法优化模型参数。train两者都是迭代细化算法;前者不依赖于多序列比对,但通常比后者慢得多。通过指定并行处理的“cores”参数,可以进一步加快 Viterbi 训练操作。训练算法的最佳选择通常取决于问题的性质和可用的计算资源。
以下代码使用 Baum Welch 算法在上一步中模拟的序列训练小珠蛋白轮廓 HMM。
-
glosf.sdPHjMlM <- trdaisn(glogbfhhjins.PHMM, sigm, metjghodh= "BaughmghWelch",
-
dk'lelt;aLL = 0.01, sejqweig;lhklkjts = NULL)

该操作需要 7 次期望最大化迭代才能收敛到指定的 delta 对数似然阈值 0.01。
序列比对
使用上述 迭代模型训练方法,可以生成高质量的多序列比对。然后以通常的方式将序列与模型对齐以产生对齐。
在最后一个示例中,我们将解构原始珠蛋白对齐并使用原始 PHMM 作为指导重新对齐序列。
-
globdins <- unaffalign(globgsins)
-
align(globinjjs, model = globinhs.PHsgMM, seqwjeighjts = NULL, resjidues = "AMINO")

请注意,列名显示了沿模型的渐进位置,以及预测插入状态已发出残基的位置(例如序列 7 的第 4 和第 5 个残基)。
参考
德宾、理查德、肖恩·埃迪、安德斯·克罗和格雷姆·米奇森。1998. 生物序列分析:蛋白质和核酸的概率模型。剑桥:剑桥大学出版社。

最受欢迎的见解