摘要:2022 年 8 月 5 日,2022 阿里云生命科学与智能计算峰会在北京望京昆泰酒店举行,圆壹智慧创始人兼首席执行官潘麓蓉博士,带来了题为《The Challenges and Future Directions of AI in pharmaceutical industry》的分享,以下是她的演讲内容整理,供大家阅览:

圆壹智慧创始人兼 CEO 潘麓蓉
美国 NIH 的 4D map 是全球制药行业的行业金标准。以小分子为例,从靶点的识别到先导化合物的发现、优化,从 early discovery 到 development 再到最终的 clinical trial,中间的每一步都已经有非常成熟的方法论、实验平台、理论指导以及监管标准。
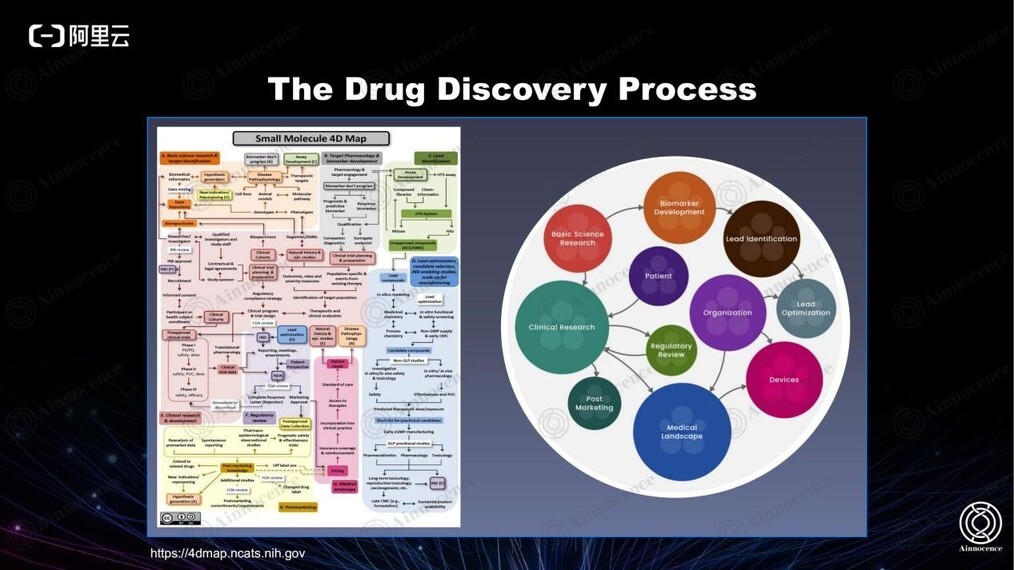
然而,该系统的数字化程度非常低,制药行业也是所有行业中数字化程度相对较低的行业。
转化医学、生物标志化合物相关的数据、临床数据、监管数据、医保数据以及临床采样和体外采样的信号数据,都需要由不同的机构和科研人员负责。基于此,制药工业想要在系统上提效,只有两个方法:第一,将整个系统进行重新定义;第二,从过去的历史数据中去掉杂音、找到信号,并用最先进的方法论取代过时的方法论。
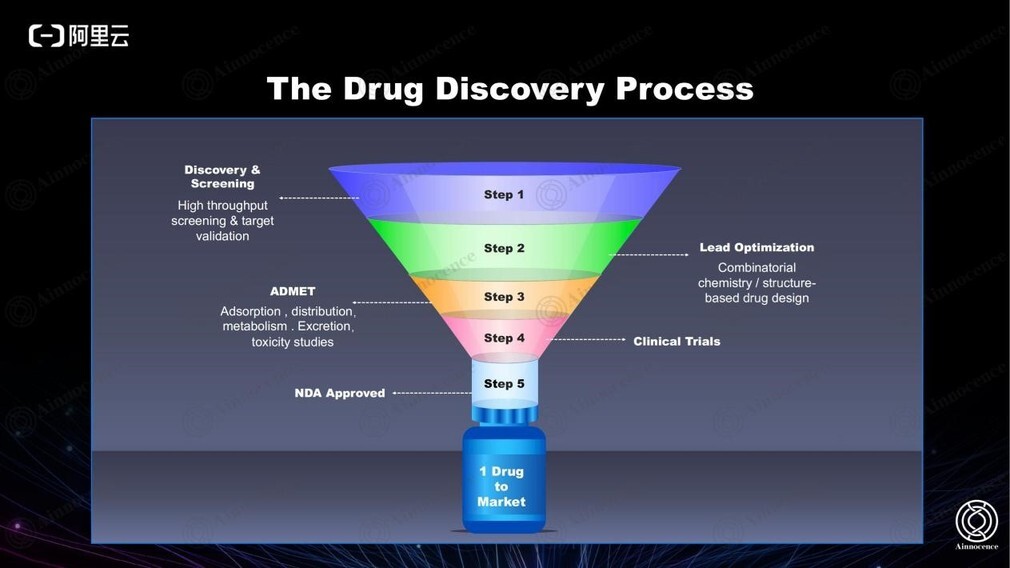
上图为药筛流程。从第一步到最后一步需要进行的实验数量决定了最终的系统效率。传统流程基本需要从 2 万个分子筛至一个分子,盲筛则基本需要 200 万个分子作为起点。而如果能实现以 100 个分子作为起点,则整个行业的投入和耗时将得到 80%以上的节省。
以上筛选流程已经沿用多年,但过去 10 年的投入产出比在逐年下降。因此我们需要考虑:如何突破现有的筛选流程?能否通过 AI 帮助提升效率?

严格意义上来说,AI 并不是一种工具,因为工具需要人来使用,而 AI 可以进行自优化,不需要人类帮助也能实现目的。在 AI 的学科定义里,它需要具备像人一样的思考和行为能力,最后还需经过图灵测试等方式的确认。
但将 AI 应用于制药行业,最大的难点在于如何为 AI 定义目标。比如制药问题上,目标可以是优化选择性,也可以是优化整体的体内药效,还可以是优化最终的适用病人群体。如果给予 AI 足够的数据,实际上它可以通过自己的办法实现目标。
因此,人需要做两件事:首先明确目标,其次明确需要喂给 AI 什么样的数据和规则,最终由 AI 负责实现目标。
人工智能本身是一个交叉学科,而制药也是涉及到生物化学、细胞生物学、生理学等多维度信息的学科。如何将众多庞大的学科体系进行高效地融合,是我们面临的最大挑战。

上图涵盖的数据基本涵盖了制药行业所有计算的输入。QM(量子力学)、DFT(Density Functional Theory,密度泛函理论)、Molecular Mechanics(分子力学)和 Molecular dynamics(分子动力学)是纯物理的方法,DFT 和 Molecular Mechanics 里也存在一些实验参数用于进行校准,而 QM 完全只取决于输入的分子的原子组成。他们在不同的精度进行计算,但精度和准度是完全两个不同的统计学参数,我们不一定需要最高的精度,但是需要最高的准确度,这样对于下一步的判断才能更完整。
而此前的方法论或多或少存在局限。比如 QM 计算的是电子精度,只能在材料和一些小型溶液化学体系里进行计算。想要扩到生物体,则需要做更多的近似和牺牲一定的精度,因此有了 DFT 方法。分子动力学方法相当于借用一些经典力学和经验参数,模拟量子力学的输出,可以将计算尺度拉到单蛋白的程度,精度从电子省略到了原子。
但是,后续需要对蛋白之间的相互作用进行计算,以及更高的体系比如细胞里有 4200 万个蛋白,如果使用 MD 进行计算,则全世界的计算机加起来都无法实现。人体需要计算生理学的结果,如果从原子开始,需要进行 4200 万*30 万亿的计算才能真正从分子层面映射到人体。受限于计算能力,从分子动力学之后,基于原子为单位对生物学的模拟随即陷入困境,且不论基于原子为基础的 3D 结构本身解析的精准程度。而随着信息学的介入,我们又看到了希望的曙光。
信息学是基于信号的读取,信号可以分为两层:一层是分子本质的信号,比如蛋白质、DNA 、小分子等都是序列,序列是确定的、没有任何噪音的;另一层是宏观层面,将分子放到体系里,可以观测到电信号、荧光信号得到各种对生物事件间接的理解。
得益于信息学的手段,过去 40 年前,化学信息学和生物信息学得到了长足发展。在此之前,我们只能用一些简单的统计学方法来实现从微观到宏观的映射。而此后的多组学能够将所有物种的 DNA 进行解析,得到多层面的数据。QM 的计算复杂度大约为 O(N)4-O(N)7,N 为电子最大的体系约几百个原子;Molecular Mechanics 的计算复杂度降至 23,最大的体系约 100 万个原子,即接近一个单病毒。但是计算复杂度在统计学或机器学习的预测场景下接近于线性,因此相当于又将计算效率节省了 10 6 -10 7 倍。当前深度学习大行其道,根本原因是我们无法通过物理学模型计算更大的生物体系,需要通过历史数据的学习来换取产生这一部分数据层投入的算力和实验资源。
DNA 是静态的,因为 DNA 的序列一般不会有太多变化。而生物是动态的,RNA、蛋白质和代谢的测量会伴随人的年纪、饮食、身体状况而动态变化。此外,当前对生命的过程模拟,从单原子角度而言大概只能达到微秒级,酶反应也大约为微秒到毫秒级,因此无法实现真正的过程模拟。而借助信息学,我们可以实现端到端的黑盒子模拟,即端到端模拟。
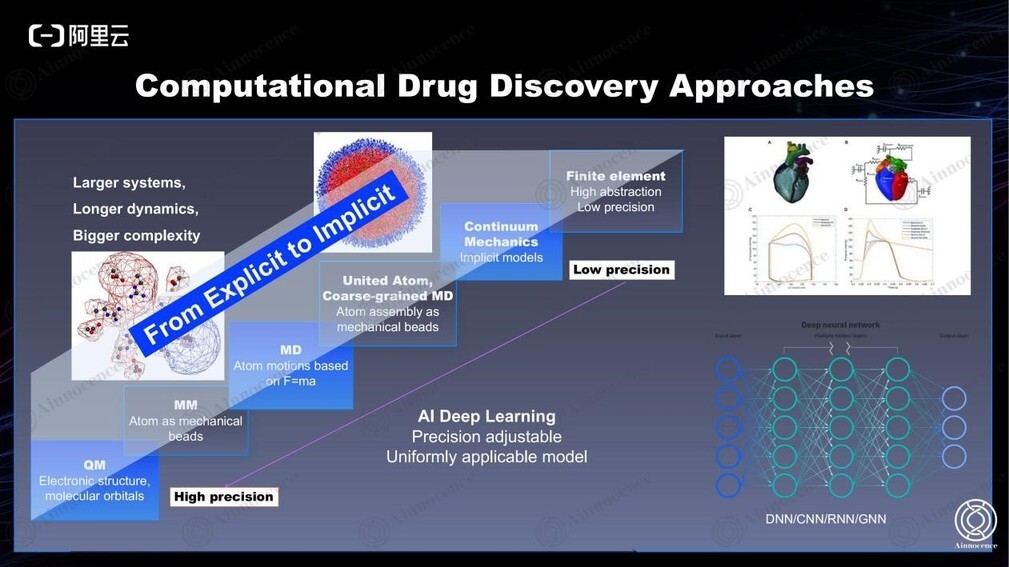
物理学家不断地简化物理公式和计算复杂度,使得最小的药物分子到体系观、不同的理论基础都可以从物理层面进行模拟。但这也意味着需要重新计算实验条件,重新发展单独的工具和物理范式,而这是一种比较笨拙的方法。我们期望能够找到精度可调、通用的模型,可以用同一个模型来解决所有问题。
深度学习就是我们的第一次尝试。只需要每个维度的数据足够多,即可用黑盒子来预测每个维度的问题,不需要考虑底层的物理原理。深度学习也在过去的实践中被证明非常有效,但它依然不是最完美的,因为它对数据过于依赖。
我们更期望的完美方式是找到一个通用型的、动态的、跨多尺度的数学公式,能够从根本上观察生物学,并且不依赖任何数据。

上图为具体的数据公式。传统的一个小分子在 QM 计算一个 GPU 大概花费几个小时至几天(取决于具体任务),FEP 大概为一天,Docking 为几分钟。而机器学习场景下,在一个 CPU 上计算几千到几百万分子只需一分钟。

上图为阿里云上测试的若干算力。QM 计算几个氨基酸的互相作用,一个 CPU 大概需花费半小时。MD 预测大型的膜蛋白每纳秒的行为需几个小时,而微秒或毫秒级所需时间则需乘以 103或 106。深度学习模型经过训练,预测所需时间更短,一个小时即可实现百万级的筛选。
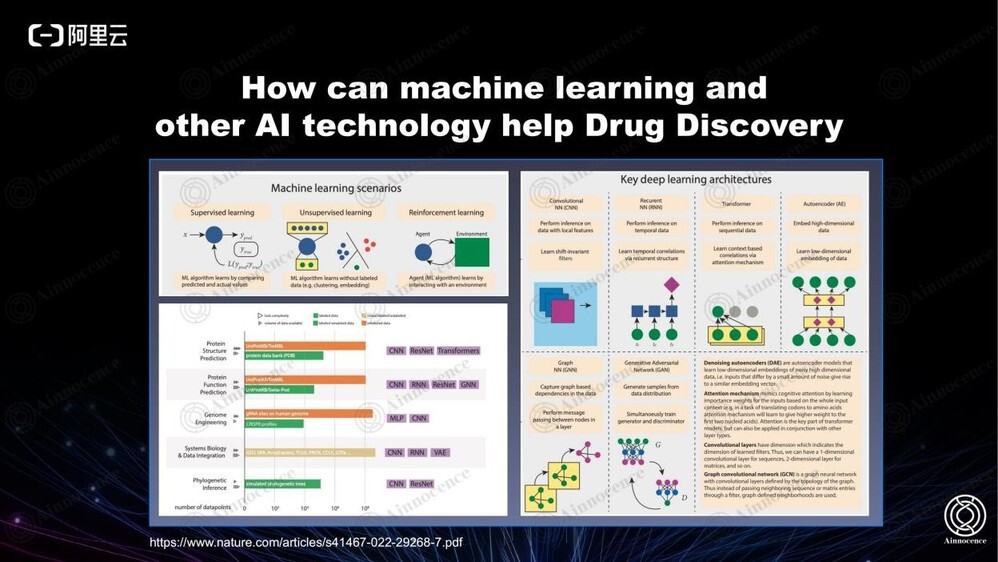
机器学习已经广泛应用于制药领域,比如蛋白质的结构预测、功能预测、基因编辑、系统生物学以及更大生理性多组学等。而最终的瓶颈在于对生物大数据的理解和清洗。
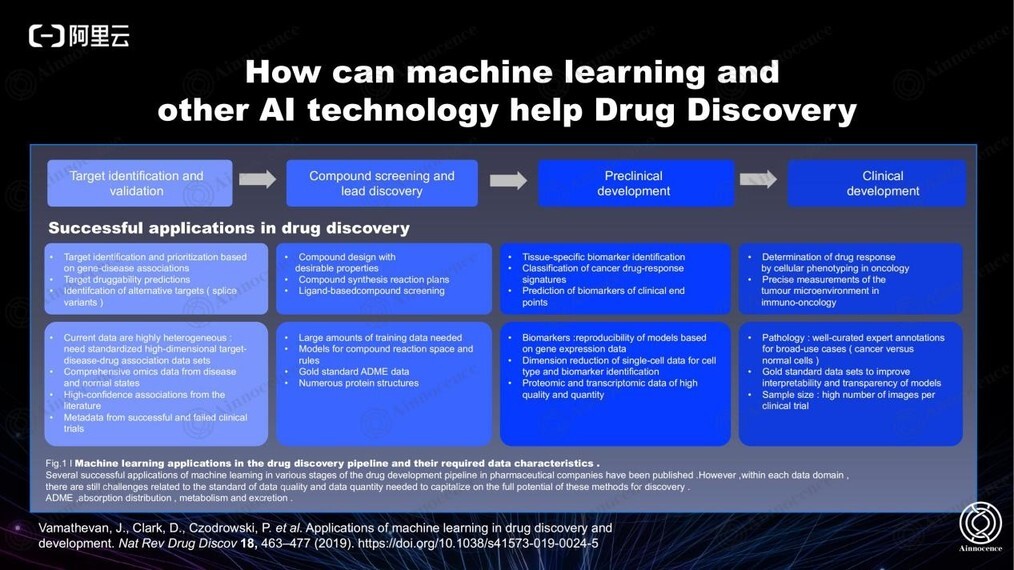
药物领域的 AI 发展主要分为上图四个阶段,到目前已经拥有完善的数据驱动方法。一直以来,我们都希望能够将整个流程里的所有数据全部打通,得到最高效的方法。

那么,从学科层面还值得继续突破的方面有哪些?

我们利用 AI,并不是只希望它做得更快,而是希望它做得更好,能实现一些人类无法突破的挑战。AI 能超越人的两个方面在于:
第一,它不需要休息,而且可以有几千个 AI agent 同时做一项工作,这是能力上的突破。
第二,AI 对于世界的认知是多维的,人只能从 3D 维度以及时间维度来认知世界,而 AI 可以在几千个维度或一维、零维这样人类无法认知的维度下认识世界,然后获得更好的答案。
制药领域存在一个很有意思的现象:二维的认知与一维的认知完全相反。如上图,PK 是影响生理指标的重要因素,不同情况下它会存在巨大反差,从人的角度看它们可能非常相似,但 AI 可以从二维以外的一些维度识别到更大的区别。
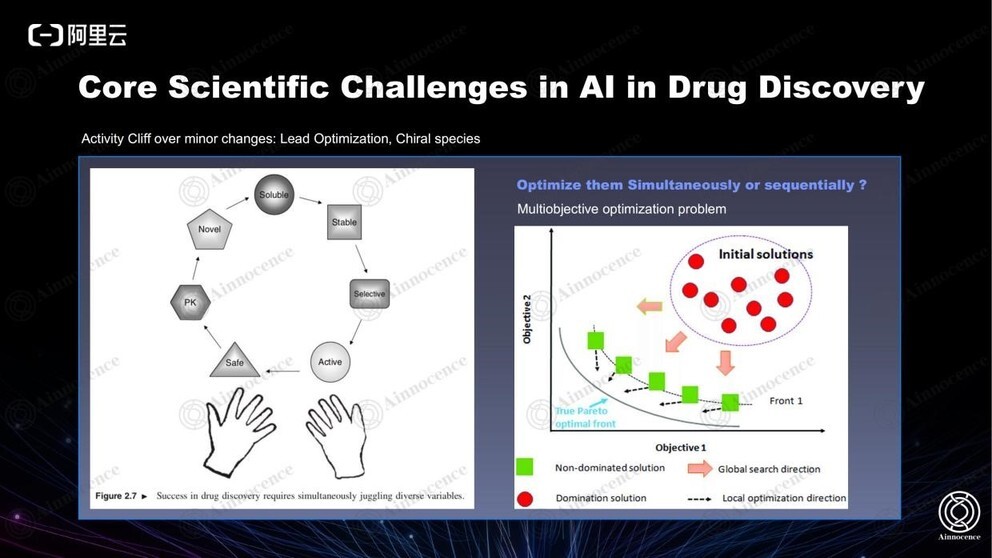
此外,专家进行优化,往往一次只能在一个维度上优化一个问题,因此一个项目会产生无限多的迭代。而如果采取人工智能最典型的 Multiobjective optimization 多目标优化方式,可以一次从多个维度实现多种优化。在过去的实践中我们已经验证,使用 AI 比如在 30 个维度里同时打分再做实验相较于人工思考再做实验的命中率要高很多。因此我们也坚信,在此领域,AI 能够比专家做得更好。
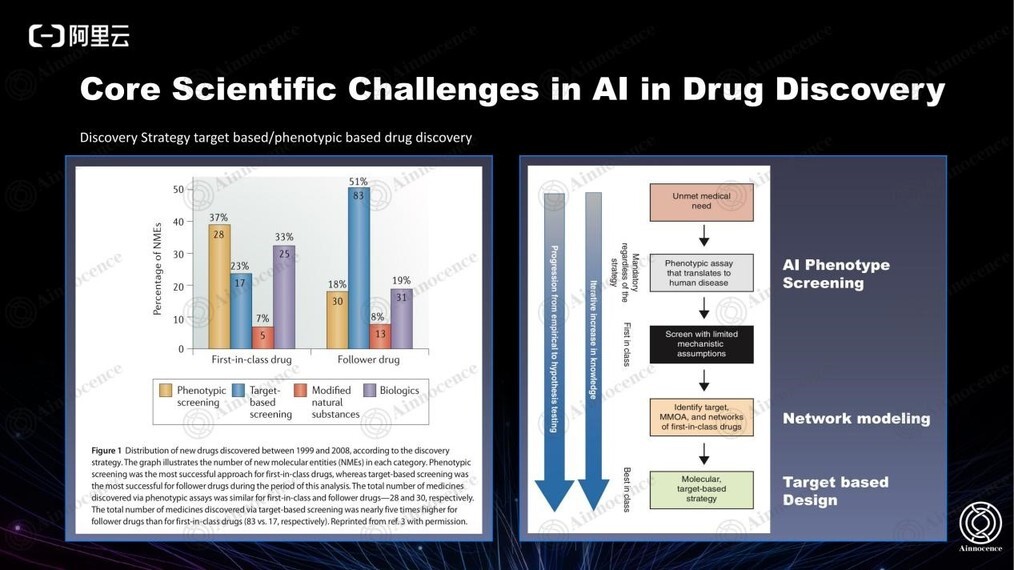
新项目一般从表型筛选开始,从表型直接预测潜在的假设,将涉及到黑盒子问题,而这正是 AI 擅长之处。过去大部分原创新药都属于 Phenotypic Screening,而大部分 Follower drug 属于 Target-based Screening。
AI Phenotype Screening 已经进行非常多尝试,比如我们过去在 GHDDI 曾对 3000 个 cell based assay 逐一进行了 AI 模型的建立,然后进行 retrospective 和 prospective 两种大规模验证,最终发现过去 30 年的数据里,只有 5% 的数据能够基本接近真实的 cell-based 结果。但这已经是一个不错的结果,至少证明了该数字保持着增长的趋势。

合成问题一直是小分子药物的瓶颈。而 Science 杂志的相关文章表明:AI passed the Turing test,意味着天然产物的全合成路径都已可预测。只是合成问题的瓶颈并不在于路线预测,而在于反应条件预测。
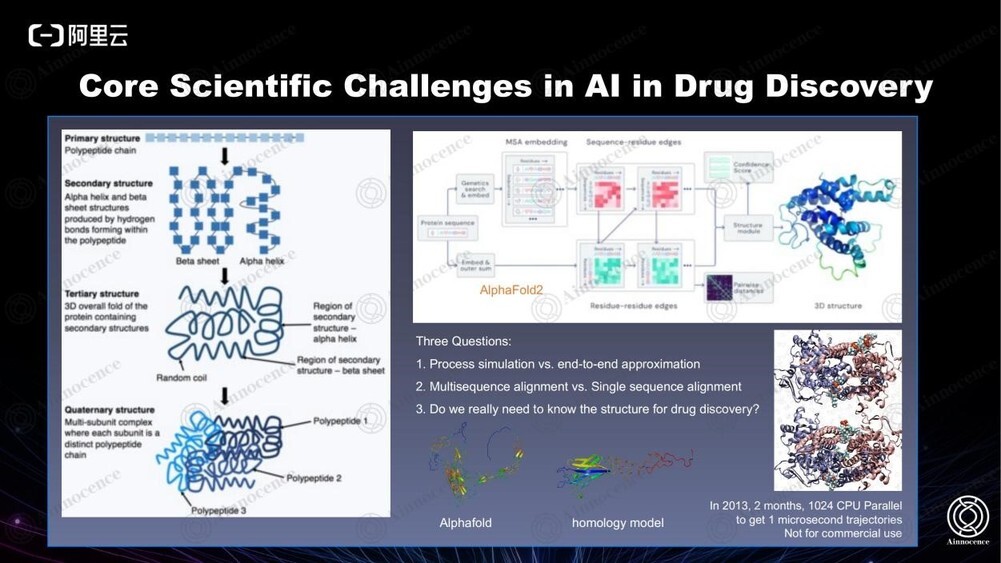
AlphaFold 饱受关注,它被认为是划时代的壮举。但我们需要先确认三个问题:
第一,制药领域是否需要知道结构?正常的 Biology discovery 可以直接在细胞上筛选或直接纯化蛋白筛。已知的只是序列和 binding affinity, 不需要过程模拟,但过程模拟的好处在于可以对一些关键位点进行改造。
第二,AlphaFold 预测的结果和传统的同源建模相比,传统的同源建模在有已知模板的情况下表现更好。其中涉及 AlphaFold 里深层次算法的 flow 使用了 Multisequence alignment,是借用其他所有物种的所有蛋白质 family 的信息去预测高等生物的信息,而这在很多核心区域会出现问题。如果是传统的同源建模一般是更接近的物种或者同一物种的同一个蛋白组族,在已知的模型上即可直接预测,因此在真实的制药过程中,传统的同源建模置信度更高。而针对没有模板的蛋白,则需要采取其他办法。
最后,我们采取的办法是直接从一级结构去预测生物活性,完全跳过了 structure biology 的过程,也就避免了这一过程中的误差。
2013 年,我曾花费两个月调用了 1024 个 CPU,得到约一微秒的膜蛋白,磷脂双分子层,小分子三种组分的 simulation,当时已经是全球最大的可计算膜蛋白体系,涉及上百万原子。而在当今的超级计算机同等硬件配置下,以上时间花费可减少至 2-3 天,但这也仅仅是 30 倍的增长,意味着真正系统性地计算动态过程依然非常困难。

因此,我们必须全方位利用 Data Driven AI 模型。通过下面这一链接,可查看这一篇滚动更新的 review 相关内容,里面提供了解决 data limitation 问题以及如何建模等方案。
相关链接:http://greenelab.github.io/deep-review/

生物大数据里的噪音非常多,如何从噪音里提取信号、集成干净的数据集也尤为重要。业内提供了非常多方法论层面、工程层面以及算法层面的解决方案,比如 Multimodal 方法,如果一个尺度上的数据量很少,则可以从其他尺度上迁移,比如 multi-task 方法,如果一个靶点的数据很少,则将其 family 或相似的所有 pocket 数据都找出来用于做迁移学习,以弥补其数据的限制。
最有用的 AI 模型一定是泛化能力很强的模型,一定能够从已知的事物预测未知的事物,这才是最有意义的 AI。因此,从根本上来说,迁移学习(transfer learning)的方法最为有效。
如果要做 target specific 预测,专家只需反馈少量结果或几个到几十个数据,即可进行 fine-tuning ,而后一般只需进行五轮以内的主动学习即可达到想要的结果,效率远远优于此前的盲筛。
另外,生成数据一般有三种方法:
第一,从现有数据里挖掘,我们曾经汇总了全球所有的商业数据库以及 100 +开源数据库,最后淘汰掉了 95%的数据,这也属于对历史的重新审视。
第二,自己做实验,有针对性地补足一些数据,需要明确数据的化学、生物空间分布,以最少的数据点推动最优的模型表现。
第三,模拟数据,比如 QM 的计算最准,则先用物理的底层采样,最后用这些数据去换已经耗费的算力,无需再重新进行计算。
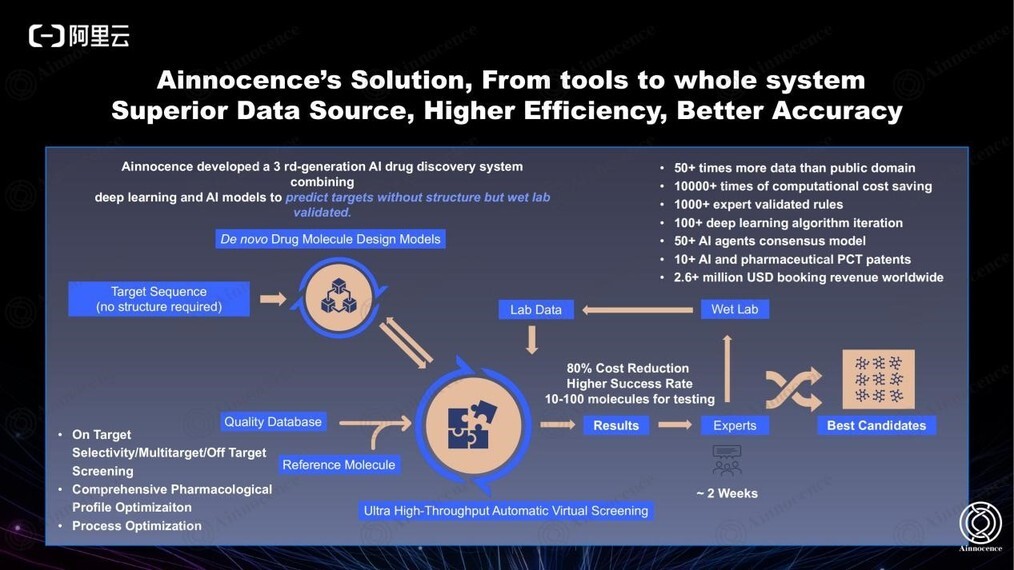
当前我们圆壹智慧的一体化解决方案如图有一个抽象的展示,具体内容参见官网,从 target 序列开始,在几个小时内通过几十个 AI 模型同时打分,可以 propose 10-20 个新分子,基本只需 2-3 轮、在 100 个分子以内即可得到目标化合物。
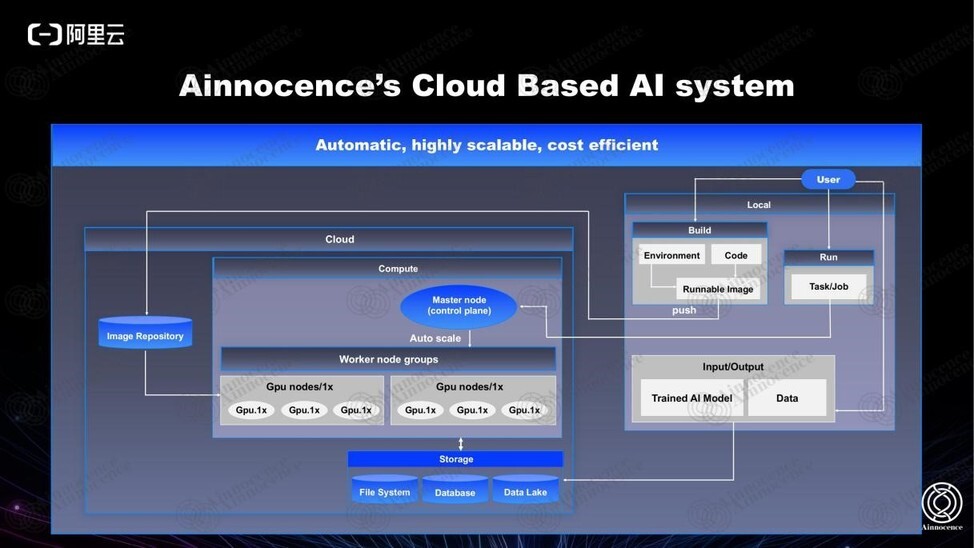
另外,在算力方面,我们从 training 、调用、 GPU 和 CPU 的分配等方面都做了非常灵活的方案,已经是一个成熟的自动化平台。

今年 6 月,圆壹智慧在生物国际大会(Bio International)上首次发布了多目标 AI 模型,对于生物药、化学药以及核酸药都提供了自动化设计的能力,并且与全世界的多家 CRO、CDMO、药企都有紧密合作,公司成立一年至今已获得 300 万美金订单。
在未来,我们也希望化学药、生物药(核酸药,蛋白药,细胞治疗)等以及各医疗产业链能够在多目标 AI 模型的加持下,更加高效地解决临床的问题。我的分享就到这里,谢谢大家。
点击这里,观看嘉宾在本次峰会的精彩演讲视频。