首先在linux 里配置conda
下载
wget https://mirrors.tuna.tsinghua.edu.cn/anaconda/archive/Anaconda3-5.3.1-Linux-x86_64.sh
chmod +x Anaconda3-5.3.1-Linux-x86_64.sh
bash Anaconda3-5.3.1-Linux-x86_64.sh
安装完毕,如果忘记选择yes,敲conda命令报错“command not found" 加上source /root/anaconda3/etc/profile.d/conda.sh
conda env list 得到 /root/anaconda3
export PATH=~/anaconda3/bin:$PATH
不然全局无法使用conda命令,(但是重启putty好像就不管用了,还不清楚原因)
vs code可以不安装
安装tree命令,yum install tree
tree -af 可以查看树形文件结构
snakemake是纯python的任务流程工具(基于python3),以前商业环境用过control-M
https://snakemake.readthedocs.io/en/stable/
首先做个变异检测,就是和标准的序列做对比,有点类似于代码的compare,用过Beyond Compare或svn和git的筒子们应该很熟悉了,
但是基因序列是个非常大的序列文件,在linux也没有windows那样简便的图形操作见面,而且,这种对比工作是大量重复的,需要脚本化。
cd snakemake-snakemake-tutorial-623791d7ec6d
conda env create --name snakemake-tutorial --file environment.yaml
--------------------------------------------------------------
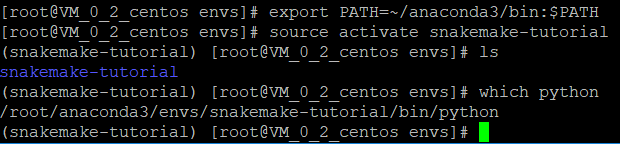
export PATH=~/anaconda3/bin:$PATH
source activate snakemake-tutorial
--------------------------------------------------------------
# 退出当前环境
source deactivate
这里使用到Samtools工具,具体使用方法可以参考https://blog.csdn.net/g863402758/article/details/53081342
他是一个用于处理sam与bam格式的工具软件,能够实现二进制查看、格式转换、排序及合并等功能,
结合sam格式中的flag、tag等信息,还可以完成比对结果的统计汇总。同时利用linux中的grep、awk等操作命令,
还可以大大扩展samtools的使用范围与功能。
conda install snakemake

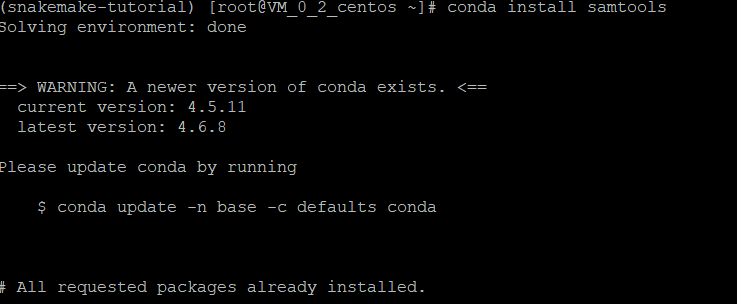
conda install samtools
bowtie2和samtools都是对比工具,bowtie2暂时没安装,安装方法先记录下
sudo wget https://jaist.dl.sourceforge.net/project/bowtie-bio/bowtie2/2.3.4.1/bowtie2-2.3.4.1-linux-x86_64.zip
unzip bowtie2-2.3.4.1-linux-x86_64.zip
vi /etc/environment
添加 bin 目录的路径,并用 : 隔开
source /etc/enviroment 使配置生效
开始写job脚本
rule bwa_map:
input:
"data/genome.fa",
"data/samples/A.fastq"
output:
"mapped_reads/A.bam"
shell:
"""
bwa mem {input} | samtools view -Sb - > {output}
"""
期间一直出一个错误,说Command must be given as string after the shell keyword
运行snakemake -np mapped_reads/A.bam检查一下是否会出错
执行这个job,把-n去掉
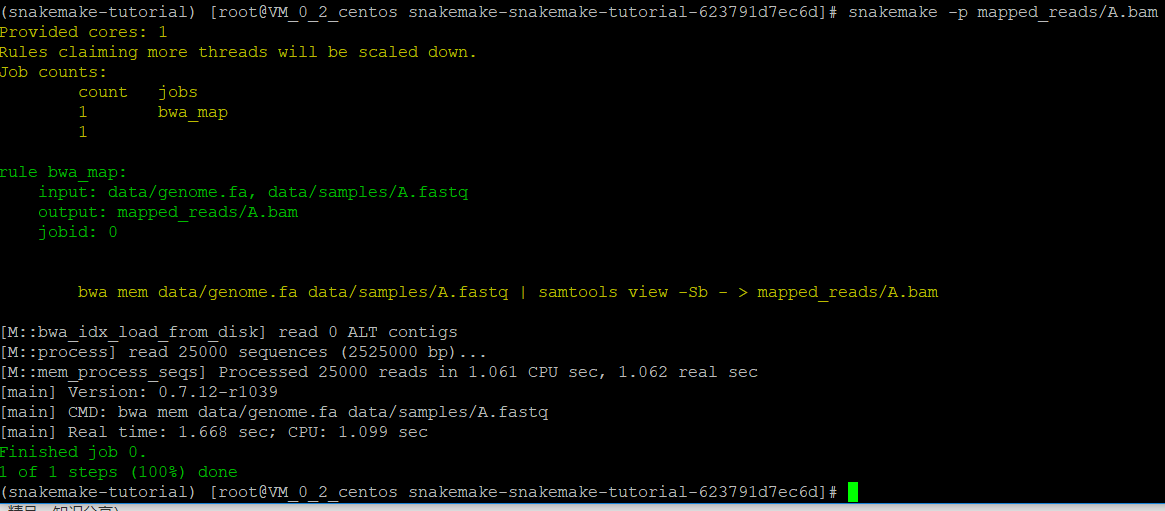
可以看到,生成了A.bam文件

rule bwa_map:
input:
"data/genome.fa",
"data/samples/{sample}.fastq"
output:
"mapped_reads/{sample}.bam"
shell:
"""
bwa mem {input} | samtools view -Sb - > {output}
"""
将A改成{sample},在输入命令的时候加上你的参数,自动匹配上了,(注意此时文件夹貌似只能有一个脚本文件),cp了一个好像报错了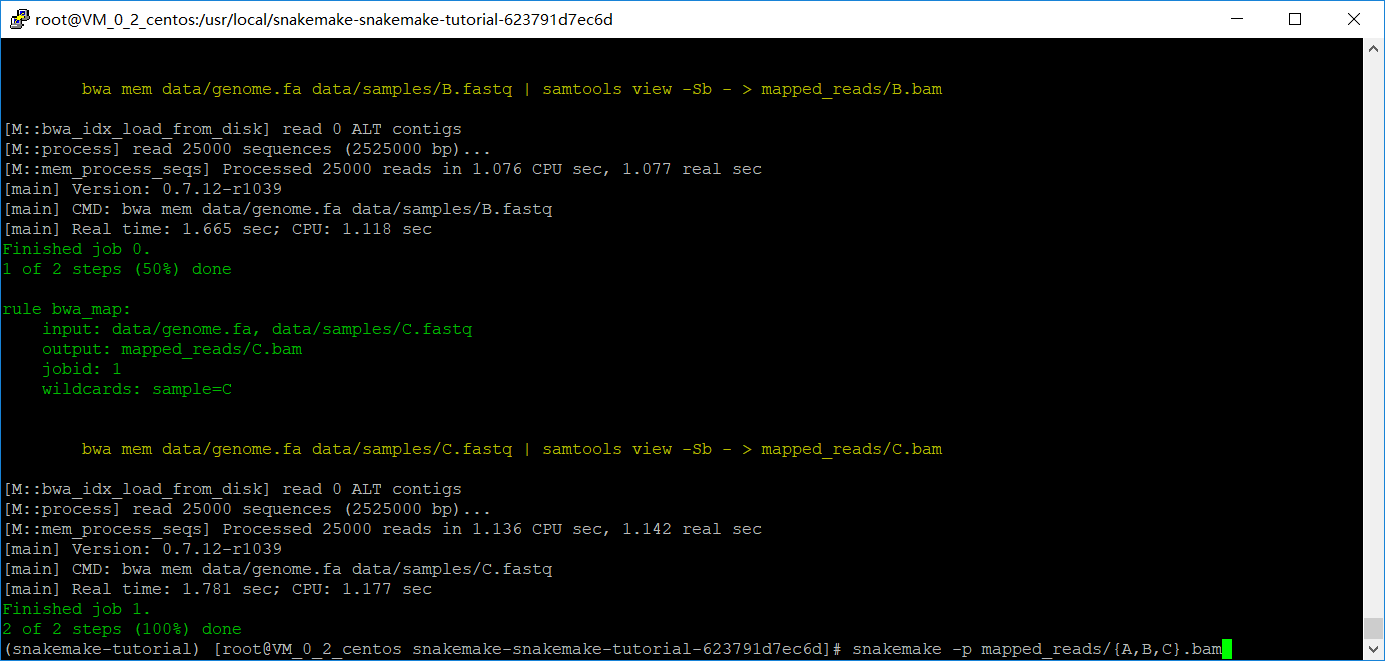
接下来,要做排序了,代码最后一起贴

可以使用dag选项和dot命令对“规则的执行和依赖关系”进行可视化,
snakemake --dag sorted_reads/{A,B,C}.bam.bai | dot -Tpdf > dag.pdf 这个命令好像会报错
snakemake --dag sorted_reads/{A,B,C}.bam.bai | dot -Tsvg > dag.svg
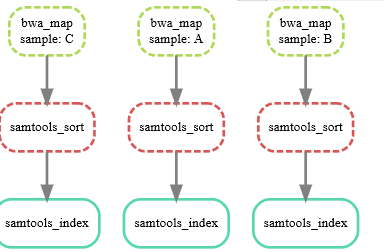
整合之前的BAM文件,做基因组变异识别
SAMPLES=["A","B","C"]
rule bcftools_call:
input:
fa="data/genome.fa",
bam=expand("sorted_reads/{sample}.bam", sample=SAMPLES),
bai=expand("sorted_reads/{sample}.bam.bai", sample=SAMPLES)
output:
"calls/all.vcf"
shell:
"samtools mpileup -g -f {input.fa} {input.bam} | "
"bcftools call -mv - > {output}"
其中expand是自动匹配变量求文件路径的语法糖
检查一下,snakemake -np calls/all.vcf
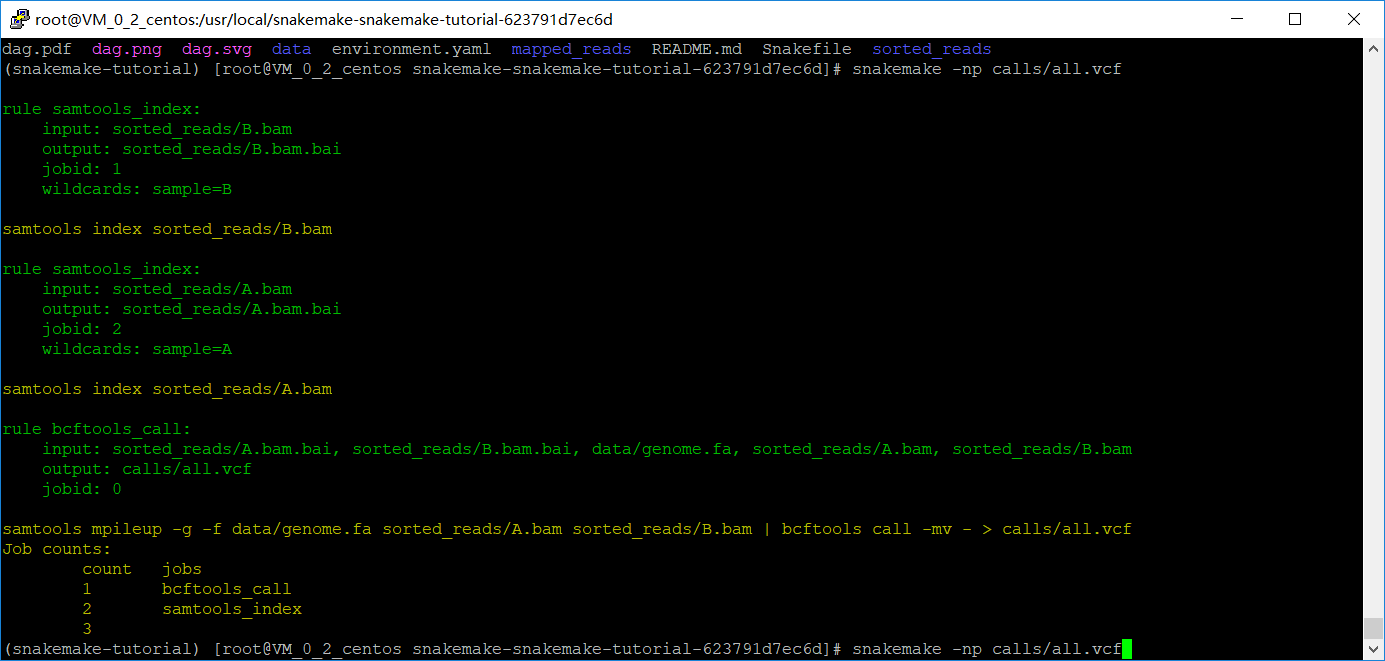
最后出report,以上都是在规则里执行shell脚本,snakemake的一个优点就是可以在规则里面写Python脚本,只需要把shell改成run,此外还不需要用到引号。
测试一下,snakemake -np report.html
画出流程图
snakemake --dag report.html | dot -Tsvg > final.svg
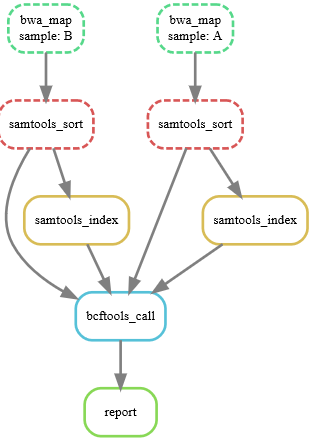
执行一下:snakemake -p report.html
可以看到生成了报告文件

到此,还有
rule all:
log:
多线程thread:
-j 指定cpu核心
params:
加载configfile: "config.yaml"
这几个功能没有操作,留个以后有空再处理
最后,在新建一个snakemake项目时,都先用conda create -n 项目名 python=版本号创建一个全局环境,用于安装一些常用的软件,例如bwa、samtools、seqkit等。然后用如下命令将环境导出成yaml文件
conda env export -n 项目名 -f environment.yaml
以后再部署的时候,
只需要conda env create -f environment.yaml
这个过程类似于ghost系统,或者打包虚拟机类似
参考了以下网址,感谢!https://www.jianshu.com/p/8e57fd2b81b2
http://pedagogix-tagc.univ-mrs.fr/courses/ABD/practical/snakemake/snake_intro.html