在生物信息分析中,经常会遇到这种情况,当数据量过大的时候会超出我们服务器的最大资源配置,导致有些软件或者我们自己写的脚本执行不过去,这时我们通常使用的办法就是将输入文件拆成很多份去依次执行,最终将结果文件合并。虽然使用python等语言可以实现拆分文件,但是既然已经有了轮子,我们就没必要去造了,这时我们就用到了linux命令split。
在linux中输入 split --help 我们可以看到它的参数如下:
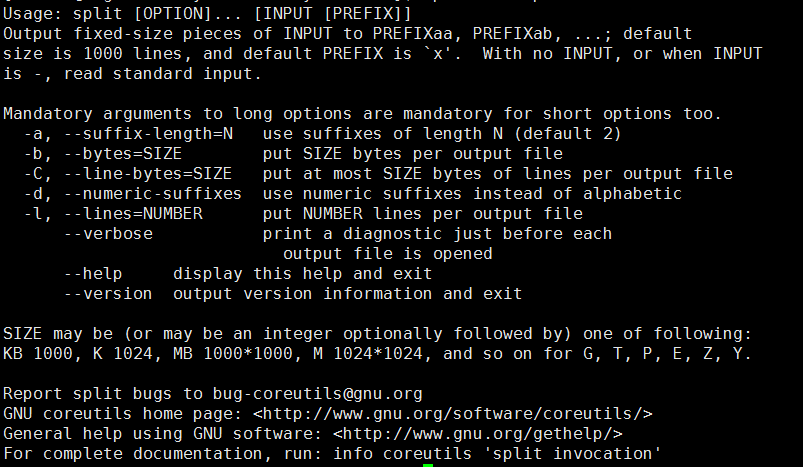
我们现在来看看它具体的参数该怎么用:
split支持自定义输出文件大小和输出文件行数两种模式,此外还可以定义每一行最大的值。
-l 按输出文件行数 : split -l 200 input_file out_file
-b 按输出文件大小 : split -b 200 input_file out_file(注意-b后边跟的数字需要换算成字节数)
注:out_file为输出文件的前缀
另外,还有 -d 和 -a 两个选项:
-d 如果加上-d则后缀为数字,不加则默认为字母
-a 默认为2,意思是后缀的位数,这个是根据你分出来的文件个数决定的
例如:

可以看到,-a为4的话out后边的位数就是4位,以此类推,设置为3就是3位。
Tips:
在拆分文件之前我们可以先利用命令 wc -l 计算一下文件总行数,然后根据我们需要分成多少个文件去简单算一下每一个文件需要多少行,会方便一些。