pindel官网:http://gmt.genome.wustl.edu/packages/pindel/
001、基本用法
pindel -i simulated_config.txt -f simulated_reference.fa -o xxxxx -c ALL
其中 -i参数用于指定配置文件;
-f参数用于指定参考基因组
-o参数用于指定输出文件的前缀
-c ALL表示所有的染色体进行分析
注:参考基因组文件, bam文件均需要提前使用samtools软件进行构建索引
002、 simulated_config.txt配置文件的格式, 如下所示:
simulated_sample_1.bam 250 SAMPLE1 simulated_sample_2.bam 250 SAMPLE2 simulated_sample_3.bam 250 SAMPLE3
第一列使用要分析的bam文件, 第二列指定长度,写个大概的值即可, 第三列指定样本名称
003、生成的文件类型:
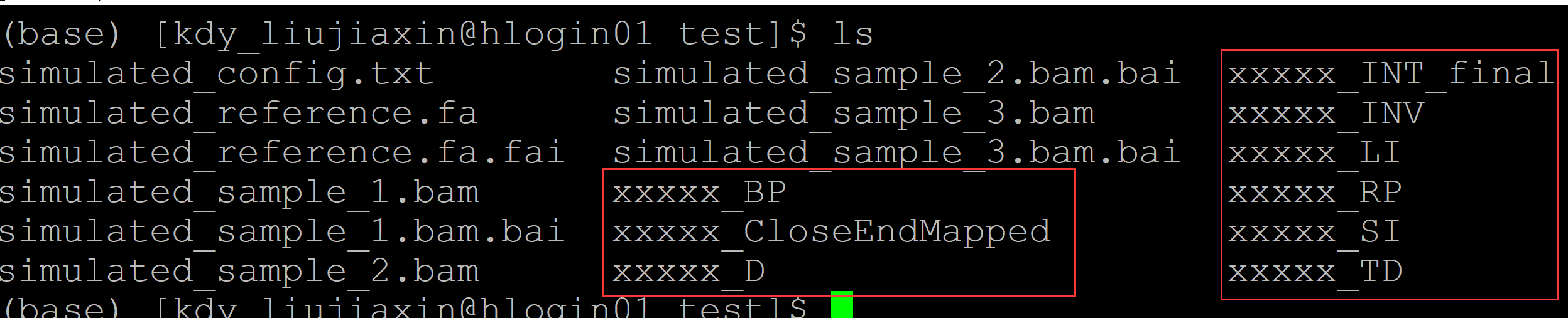
xxxxx_D:表示deletion
xxxxx_INV: 表示inversion
xxxxx_LI:表示long insert
xxxxx_SI:表示small insert
xxxxx_TD:表示tandam duplication
xxxxx_BP:表示unassigned breakpoints 没有分到上面任意一种类型剩下来的断点
004、将结果文件转换为易读的vcf文件
pindel2vcf -p xxxxx_D -r simulated_reference.fa -R simulated_reference -d 20220925 -G -v yyyy.vcf
-p:指定要转换的文件类型:
-r:指定参考基因组:
-r:指定参考基因组前缀
-d:随便写
-G:指的是转换为尽量是和GATK兼容的格式
-v:指定输出为vcf文件
参考:
001、https://www.cnblogs.com/lmt921108/p/13964733.html
002、https://www.jianshu.com/p/1815de1090d3