1)blast产生背景
双序列比对可以采用是基于动态规划算法的Needleman-Wunsch(NW)和Smith-Waterman algorithm(SW)算法,虽然精度高,但计算消耗大。当与数据库比对的时候,该算法就显得不切实际。因此TASTA,blast采用启发式算法使得通过大幅度丢失灵敏度来减少运行时间。与FASTA软件相比,blast通过把搜索限制在狭隘的矩阵对角线条带上,来改进FASTA进行数据库搜索的速度。
2)blast的大致原理
blast 程序首先查询query序列的所有子序列,储存在哈希表中。收索数据库中所有与子序列精确匹配的序列,作为种子,向两个方向继续延伸每个精确匹配。期间不允许有空位和错配的情况。然后在限制性区域内;连接延伸的匹配序列,期间允许空位和错配,比对分值要大于设定的阈值。阈值越大,需要匹配的计算越小,软件计算速度越快。仅仅对对延伸匹配进行连接的区域(限制性区域),而不是整个矩阵,是blast 相对于其他算法速度提高的关键,是以牺牲对角线带以外的任何匹配信息为代价,因此并不能确保query序列与数据库比对结果是最优的比对结果。
3)blast的格式解读
blast 输出格式:有18个格式,其中常用的outfmt 6
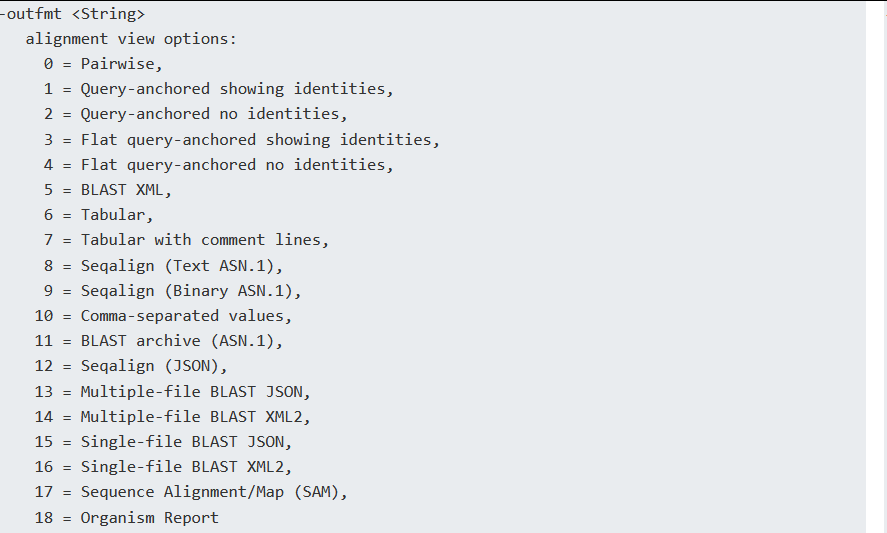
其中,格式6、格式7、格式10、格式17的输出条目是可以修改的。输出格式选择 6 (--outfmt 6) ,默认输出为:qseqid sseqid pident length mismatch gapopen qstart qend sstart send evalue bitscore 。
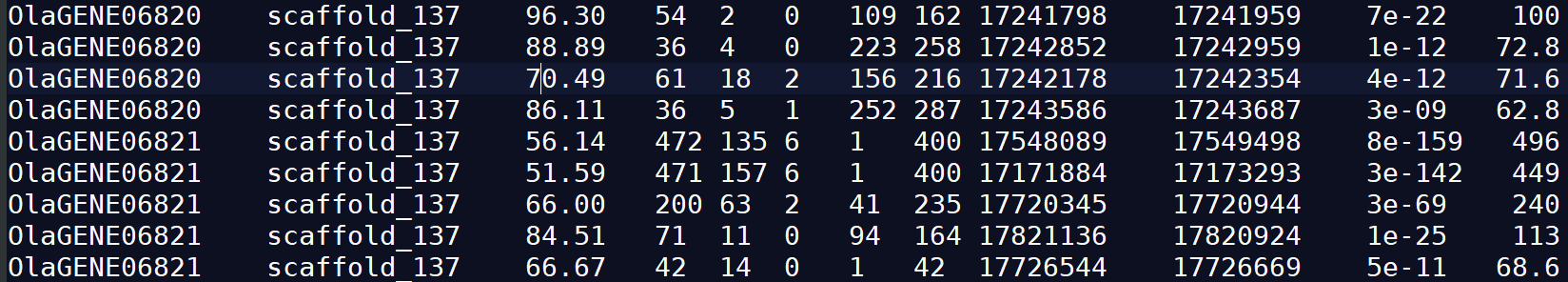
这12列对应的信息分别是:
Query id:查询序列ID标识
Subject id:比对上的目标序列ID标识
% identity:序列比对的一致性百分比
alignment length:符合比对的比对区域的长度
mismatches:比对区域的错配数
gap openings:比对区域的gap数目
q. start:比对区域在查询序列(Query id)上的起始位点
q. end:比对区域在查询序列(Query id)上的终止位点
s. start:比对区域在目标序列(Subject id)上的起始位点
s. end:比对区域在目标序列(Subject id)上的终止位点
e-value:比对结果的期望值,将比对序列随机打乱重新组合,和数据库进行比对,如果功能越保守,则该值越低;该E值越高说明比对的高得分值是由GC区域,重复序列导致的。对于判断同源性是非常有意义的几个参数。
bit score:比对结果的bit score值
如果想要获取比对物种信息 taxid ,则需要在建立索引时添加物种分类信息,同时比对时自定义添加一些输出信息,详细说明参见软件手册,示例如下:
# 建立索引 diamond makedb --in nr.gz --db nr --taxonmap prot.accession2taxid.gz --taxonnodes Taxonomy/nodes.dmp --taxonnames Taxonomy/names.dmp ## 蛋白序列比对 NR 全库 diamond blastp --db nr --out test_pep_vs_nr.out --outfmt 6 qseqid qlen sseqid slen pident length mismatch gapopen qstart qend sstart send evalue bitscore staxids sscinames salltitles --query test_pep.fa --max-target-seqs 1 --evalue 1e-5 --max-hsps 1 --threads 45 --block-size 20 --index-chunks 1 --tmpdir tmp --more-sensitive
5)参考资源
https://en.wikipedia.org/wiki/BLAST#Output
第二代测序信息处理