参考资料来源:
http://software.broadinstitute.org/gsea/doc/GSEAUserGuideFrame.html
https://cloud.tencent.com/developer/article/1426130
GSEA图的理解:
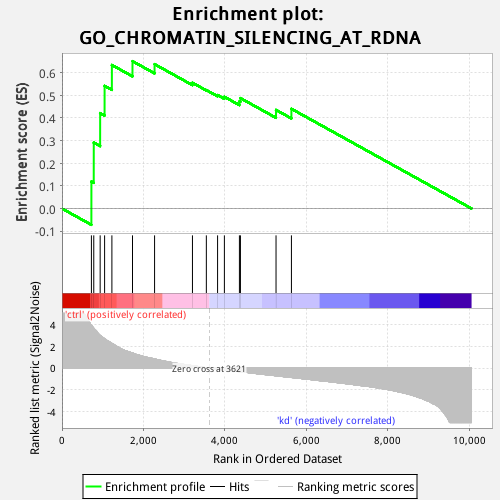
含义概述:
竖线表示:某个GO过程(比如:脂肪代谢)的基因。
我测了10000个基因在treat组和control组中的表达量。那么,与脂肪代谢相关的基因,在这两个组中是有差异表达的吗?
通过此图,可以知道这个信息。
竖线的基因集中在左端,说明这些基因在treat组中高表达。
得出结论:与脂肪代谢相关的基因,在treat组和control组中,是差异表达的。这些基因在treat组高表达。
若黑线不聚集在某端,而是散落地排列。说明:与脂肪代谢相关的基因,在treat组和control组的表达没有差异。
含义详述:
给定一个排序的基因表L和一个预先定义的基因集S (比如编码某个代谢通路的产物的基因, 基因组上物理位置相近的基因,或同一GO注释下的基因,即MSigDB数据库中的基因),GSEA的目的是判断S里面的成员s在L里面是随机分布还是主要聚集在L的顶部或底部。这些基因排序的依据是其在不同表型状态下的表达差异,若研究的基因集S的成员显著聚集在L的顶部或底部,则说明此基因集成员对表型的差异有贡献,也是我们关注的基因集。
比如:根据数据库,上图的GO过程有一个先验的gene集(即S),我也有一个自己的50000+个排序基因集(即L)中,此GO过程中有多少个基因落入我的基因L中呢?答:图中间的黑色条带表示GO过程基因落入排序基因集L中的基因。这些基因在ctrl中聚集还是sd中聚集呢?答:在ctrl中聚集。这些黑条带基因是随机分布在50000+的基因集中,还是显著聚集在50000+基因集的顶部或底部?答:聚集在顶部。
1. 图中下面灰色图的含义:http://software.broadinstitute.org/gsea/doc/GSEAUserGuideFrame.html?_Interpreting_GSEA_Results。所有的灰色图都是一样的,即结果文件ranked_gene_list_treat_versus_control_1572940764277.xls中的值。
图中的横坐标表示你输入的gene列表。
1)纵坐标含义:纵坐标表示“Ranked list metric”,GSEA官网对此的解释是ranking metric,即signal-to-noise ratio。
2)计算方法:参考下面的Signal2noise的解释。
3)ranking metric的含义:它表示基因与表型(phenotype)的关系。即:这个基因与treat相关,还是与control相关,相关的度量值是多少。即ranking metric的含义。
positive ranking metric表示与phenotype1相关,negative ranking metric表示与phenotype2相关。
表型是什么?比如,我们对给药组(treat)和正常组(control)做RNA-seq,得到每个基因在给药组和正常组中的表达值。treat和control就是表型。
主要概念 :
Signal2noise:信噪比。信号强度/噪声强度。值大于1,表示信号强于噪声;值小于1,表示信号弱于噪声。举例:gene A在treat组和和control组的表达量的比值。上图中,对该值取log,值大于0,表示在前表型中表达量高;值小于0,表示在后表型中表达量高。即:result文件中“ranked_gene_list_treat_versus_control_1572940764277.xls”的含义。
ES(enrichment score,富集得分):它是一个累积值。表示GO过程的基因集S在排序基因集L中的富集程度。(之前,我总是想不过来这一点。因为我想的是:排序基因集L中有多少个基因落入GO过程的基因集中呢?其实,应该反着想:GO过程的基因有多少个落入排序基因集L中呢?是散落在两种phenotype之间,还是富集在一种phenotype中呢?)正值ES表示该GO过程的基因集在前表型(phenotype1)中富集,负值ES表示基因集在后表型(phenotype2)中富集。
计算方法:首先,对基因集L按照Signa2noise排序(除了Signal2noise,GSEA也有其它的计算方法),得到排序基因集L。
然后,对L中的每个基因,根据这两个条件:基因是否出现在GO基因集S中;该基因的Signal2noise值。计算得到L中每个基因的Running ES值。(这两个条件体现了基因集L与phenotype的关联度)
最后,对L中的每个基因,计算累积值。即得到上图的第一个图。
在上图第一个图中,ES的最高或最低值,作为该GO过程的ES值。
NES(Normalized Enrichment Score):它用来比较各个GO过程之间的ES值。上面的ES值是一个GO过程内部的每个Gene的ES值。而,对于每个GO过程都有一个NES值。
FDR :q-value。gene sets的错误概率。(理解:该GO过程包含这些基因的错误概率)。对NES值的估计。FDR=25%表示在4次验证中,有3次是成功的。如果样本数少,用gene_set作为permutation,那么,需要更样的FDR,比如<5%.
gene set:某GO过程的基因集。
p-value:对每个gene set都有一个p-value。它表示某个gene-set的ES值的显著性。
置换检验结合GSEA说明(参考”置换检验—结合GSEA解释“的文章):
随机抽取次数(比如上文的1000),即是GSEA中提到的number of permutation。次数越多,得到的统计量的值越多,抽样分布的值越多,抽样分布的分布图更接近真实值。看真实数据在此抽样分布图上的分布时,就更准确。
P-value:
由以上可得知P-value的含义。拒绝零假设的概率、上图的末梢的面积、”GO过程在排序基因集L中富集“这一结论错误的概率。
通常,我们取P值的阀值为0.05。即:P值小于0.05的GO过程都认为在排序基因集中是富集的。
那么,既然错误率是0.05,也就是说:检测100次,有5次是出错的。如果检测1万次,有500次是出错的。也就是说,当检测次数很多时,出错的情况就会多。结合GSEA:GSEA给出的GO过程(P值小于0.01)中,100个GO过程中有5个是出错的。即:100个GO过程中,有5个GO过程在两种phenotype中是不富集的。10000个GO过程中,有500个是不富集的。当GSEA给出富集的GO过程很多时,假阳性的GO过程就很多。
适用场景:
例子:cell,2016年发表的文章(https://sci-hub.tw/10.1016/j.cell.2016.11.033),使用了GSEA做富集分析。
参考GSEA的官网说明:
Gene Set Enrichment Analysis (GSEA) is a computational method that determines whether an a priori defined set of genes shows statistically significant, concordant differences between two biological states (e.g. phenotypes).
我理解的GSEA分析所适用的场景之一:对特定的基因集,在两种phenotype中的富集程度。
举例说明:有两种类型的细胞,treated和control。想知道哪种功能的基因集在这两种类型的细胞上有差异。比如:与髓鞘形成相关的一个功能,这个功能对应一个基因集(即:GSEA数据库中的基因集),想知道与髓鞘形成相关的基因在treat组上表达,还是在control组上表达。要实现这个功能,需要知道这些数据集:treat和control组的与髓鞘形成相关基因的表达值(RNA-seq得到);与髓鞘形成相关的基因都有哪些(GSEA MSigDB的GO数据库)。
我理解的GSEA分析所适用的场景之二:对于两种phenotype,哪些gene set在这两种phenotype中有差异。
参考资料:
原文发布于微信公众号 - 生信宝典(Bio_data)
原文发表时间:2019-04-26
工具下载:
下载网址:http://software.broadinstitute.org/gsea/downloads.jsp
命令:
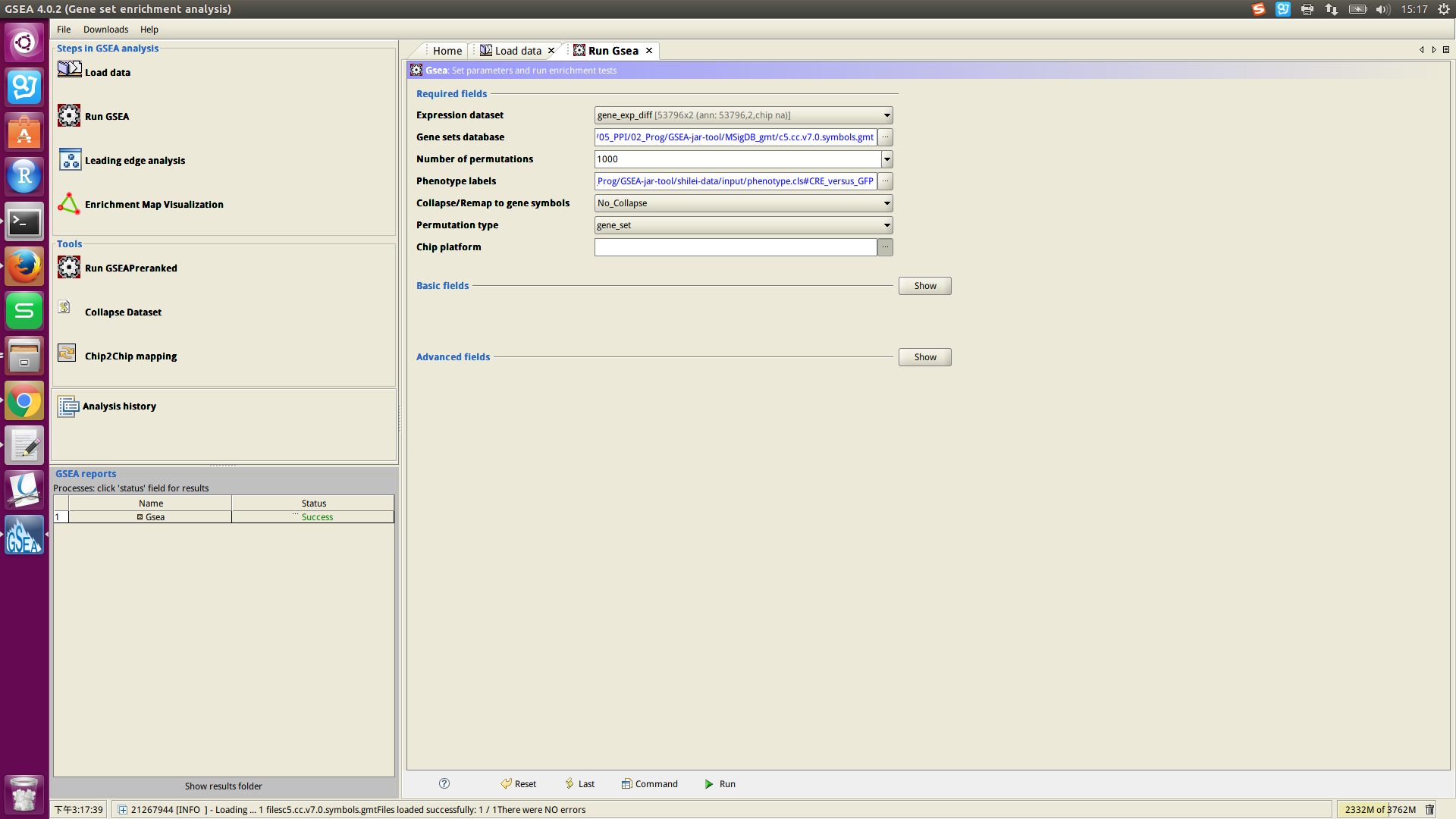
执行命令:./gsea.sh ,打开了界面版的GSEA工具。
简要步骤:
Load data,需要load:expression文件、phenotype文件、下载的MSigDB的文件。
设置Required fields的参数,点击最下面的Run。
详细步骤:
使用:
准备输入文件:
1. Expression dataset:是RNA-seq(或chipseq)得到的数据。
通常有这么几列:
例1:
NAME DESCRIPTION AML1 AML2 AML3 ALL1 ALL2 ALL3
TP53 na 681.3 638.0 665.0 240.0 587.0 737.0
例2:
NAME DESCRIPTION G C
TP53 na 681.3 638.0
注意:
1)必须用tab分隔;
2)第一行的名称必须是NAME和DESCRIPTION;
3)文件前两行必须是:
#1.2(固定的内容)
53796(gene个数,即行数) 2(sample个数,不是phenotype数。如例1中phenotype数是2,sample数是6) 参考网址:http://software.broadinstitute.org/cancer/software/gsea/wiki/index.php/Data_formats
2. Phenotype labels文件:共三行。
2 2 1 分别为:sample数,phenotype数,最后的1是固定的
# G C phenotype的名称
G C phenotype的名称
以上两个文件是需要自己准备的,是自己的的数据。
3. Gene sets database选项:
可以用GSEA ftp网站上的,也可以从GSEA 下载MSigDB的文件,load到工具中使用。
4. Collapse/Remap to gene symbols选项:
我选择的是“No_Collapse”。
5.Permutation type选项:
GSEA文档中,给出的说明是 :样本数大于7,建议用“phenotype”选项;小于7个样本的数据,建议用“gene_set” 的选项。
6. Number of permutations选项:
GSEA官网建议1000。刚开始运行时,建议10;运行成功后,再设置为1000。
术语说明:
GSEA文档中的Phenotype、sample、gene sets等的含义。
Phenotype:
sample:
gene sets:
问题:
1.error:“Read timed out!”
这个错误,gsea官网没有说明。自己查的解决方法,费劲...。
原因:Gene sets database中,我选的是website的dataset,比如:ftp/...。这样,程序运行时,去GSEA的ftp服务器寻找并使用该文件。我怀疑可能网络不好,导致读取超时。
解决:下载gene set到本地。其实,就是把MSigDB的数据文件(.gmt文件)下载到本地。下载地址:http://software.broadinstitute.org/gsea/downloads.jsp。
注意:load本地的gene set文件时,只能通过“Load Data”功能执行。我在gene set database打开的界面找了好久都没找到load文件的图标,最后,灵机一动,才想到如何load。浪费时间%>_<%
2. error:After pruning, none of the gene sets passed size thresholds.
这个错误,gsea官网有说明。
原因:下载的c5.all.v7.0.symbols.gmt文件中的gene symbol全是大写,而我提供的expression文件中的基因名是小写。
解决:将expression文件的基因名改为大写即可。
具体解决方法:awk '{print toupper($3)}’,使用shell脚本的toupper函数即可。
3.运行时间:
我输入两个sample,两种phenotype,共53000+个gene的数据,permutation选择默认值1000,选择一个bp.gmtwenjian,运行时间长,大约十几分钟。