分子模拟定义及方法
将一定数量的分子输入计算机内进行分子微观结构的测定和宏观性质的计算。按照获得微观态的方法不同,分子模拟可以分为:
1.Monte Carlo蒙特卡罗方法,MC:1.构型平均,不包含动力学部分;2.利用概率行走产生微观态。
2.Molecular Dynamics分子动力学,MD:1.时间平均,产生动力学性质;2.利用运动轨线随时间的变化来产生一系列微观态。
主要依靠牛顿力学来模拟分子体系的运动,以在分子体系的不同状态构成的系统中抽取样本,从而计算体系的构型积分,并以构型积分的结果为基础,进一步计算体系的热力学量和其他宏观性质。目的是用少量粒子的长时间平均行为,来试着取代大量粒子的瞬间平均行为。
3.hybrid method混合方法,HM
分子模拟涉及的几个基本概念
1.Simulation box(cell)模拟计算盒子或模拟胞腔:装有一定数目流体分子的研究对象,它是我们要研究的宏观体系的微缩模型。设盒子边长为L,则体积为V=L3,分子的质量为m,则系统的密度为d=Nm/L3,系统的密度=实验室测定的密度。
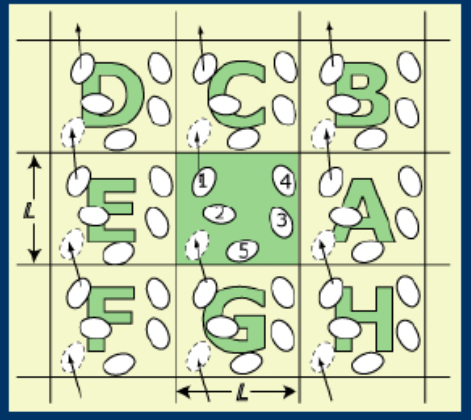
2.Periodic boundary condition周期边界条件,PBC:
在小体系中,边界效应总是很显著。在模拟中,考虑具有真实边界的对象,不切合实际(增强了有限尺寸效应;人为造成的边界会影响流体的性质)。故需要以下算法:本体系的近似——中心盒子在x,y和z方向上无限扩展,消除人为形成的边界的表面效应,保证中心盒子中的粒子数恒定,只需要跟踪中心盒子中个粒子的运动。(当某个粒子运动出模拟盒子的某一边界时,另外一个影像粒子从另一对立边界进入到此盒子中)
Fortran中常用函数:
Dble(x)*把数据x转换成双精度实数,x:I
ANINT(x[,kind])*对x四舍五入取整,并转换为实数(kind),x:R,kind:I,结果:R(kind)
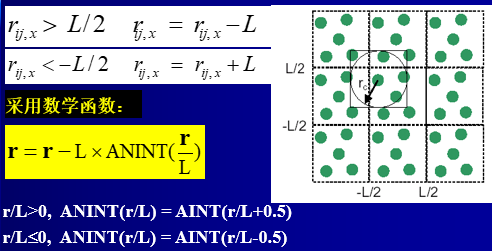
3.Minimum image convention最小影像转化原理:中心元胞中的一个粒子只与此元胞中的其它N-1个粒子,或它们的最近邻影像发生相互作用。适用条件:粒子间相互作用势能的截断距离必须不大于模拟中心元胞长度的一半。
最小影像转化原理的算法:
4.Truncating the Potential截断势能:粒子间的相互作用主要来自于截断范围内,而范围外的贡献很小,可忽略不计。(不大于最小边长的一半)
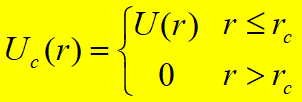
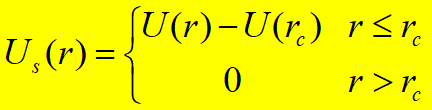
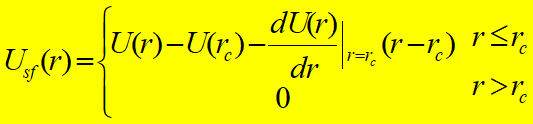
Truncated Potential简单截断势能函数: Shifted and Truncated Potential位移截断势能函数: Shifted-Force Potential位移-力截断势能函数:
势能在截断处不连续,能量不守恒 常用于MC,MD模拟中,但势能在截断处连续 常用于MD模拟中,分子间力和势能在截断处均连续
分子间力在截断处不连续且无穷大,MD运动不稳定 分子间力在截断处不连续,不为无穷大但势能在截断处连续
分子动力学基础
1.如何实现纳米尺度的材料研究与宏观尺度的实验结果的比较?分子动力学元胞+周期边界条件
要找到合适的元胞,需要对不同尺寸的元胞进行试验(收敛性试验),找到收敛趋势,得到截断值,由此得到代表性单元。(注意找收敛趋势的时候要画在同一坐标里)
元胞模型的建立:(1).材料的原子结构——材料所含个原子的空间位置,可由x射线衍射法实验获得,详细信息可通过数据库查找:American Mineralogist Crystal Structure Datebase;(2).材料原子间立场的定义——个原子间的相互作用势,包括库仑力,分子内部原子相互作用力,分子间原子相互作用力(范德华力)。定义库伦力势与分子内部及分子间原子作用力势(简单为好)
分子总能量=动能+势能(几何坐标的函数)


非键结作用:最基本的应用最多的是二体势(对势),最常用的是Lennard-Jones对势,ε是能量参数,σ是长度参数,如下:

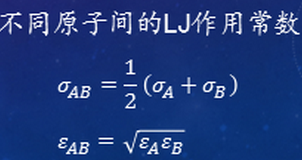
将牛顿运动定律方程式对时间积分,可以预测原子经过时间t 后的速度和位置。其数值解法:verlet方法