vegan 包是进行群落数据分析最常用的R包,其中的 specaccum 函数用来计算物种的累计曲线
首先看下官方示例:
library(vegan) data(BCI) sp1 <- specaccum(BCI, method="random") plot(sp1, ci.type="poly", col="blue", lwd=2, ci.lty=0, ci.col="lightblue") boxplot(sp1, col="yellow", add=TRUE, pch="+")
出来的结果图如下:
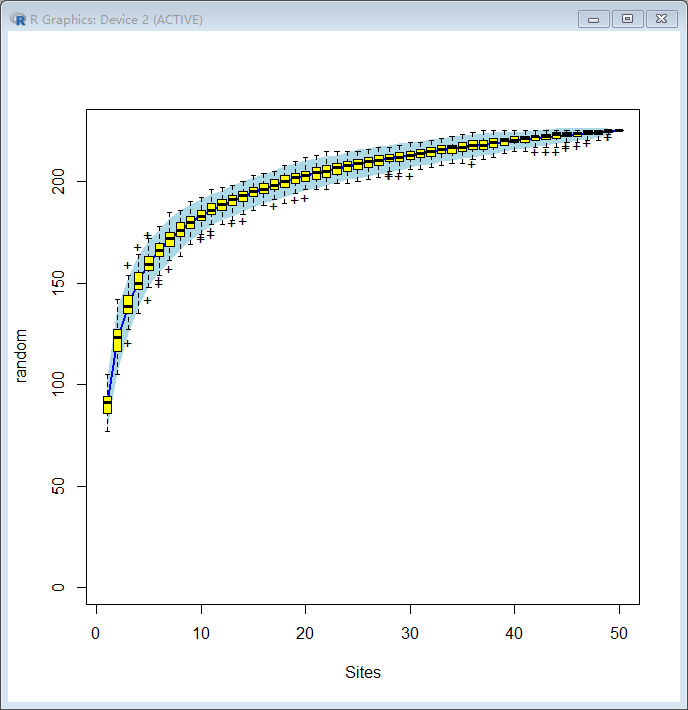
那么这幅图表明了什么含义呢?
首先看下输入数据
> head(BCI[, 1:3]) Abarema.macradenia Vachellia.melanoceras Acalypha.diversifolia 1 0 0 0 2 0 0 0 3 0 0 0 4 0 0 0 5 0 0 0 6 0 0 0
我们简单的看一下BCI这个数据,它的每一行代表了一个样本,不同样本采样的地点不同,每一列是1个物种的丰度
最终的物种累计曲线中,横坐标是样本个数,纵坐标是发现的物种个数,随着样本个数的增加,发现的物种个数也不断增加;
其实,物种累计曲线反应的就是抽样个数对物种多样性的影响;可以看到,当抽样个数较少时,发现的物种并不全面,并不能表征整个群落结构,随着抽样个数的上升,发现的物种数越来越多,也更能表征这个群落结构;
在实际分析中,什么样的情况表明我们的采样量足够了呢,主要看曲线的末端,如果曲线末端部分还呈现 急剧上升的趋势,表明抽样量不足;增加样本量,还能继续发现新的物种;当曲线末端上升趋势趋于平缓时,则表明采样量足够,