GATK处理DNA 水平的snp 经验比较成熟,而RNA 水平较少,所以可能会存在错误
目前的流程兼顾了假阳性(不是真的snp位点)和假阴性(该位点是snp,却没有检测到);后续会不断改善
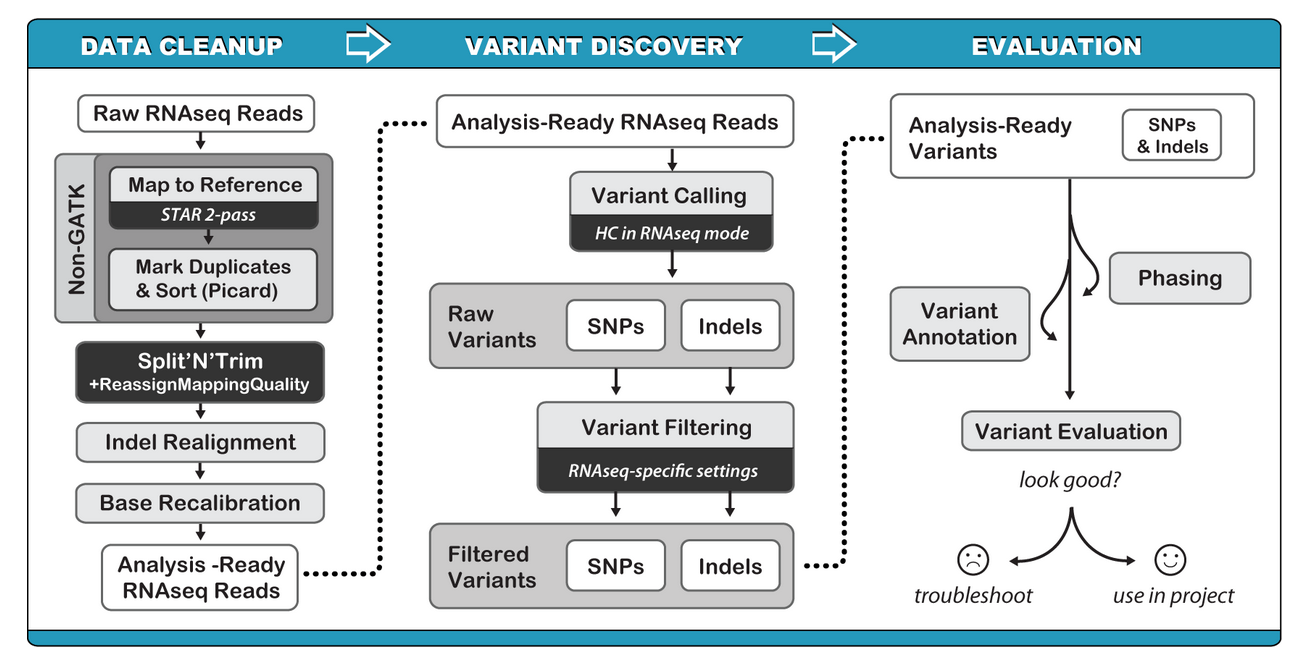
GATK SNP calling pipeline 分成3个部分:
1)DATA CLEANUP
2) VARIANT DISCOVERY
3) EVALUATION
DATA CLEANUP :
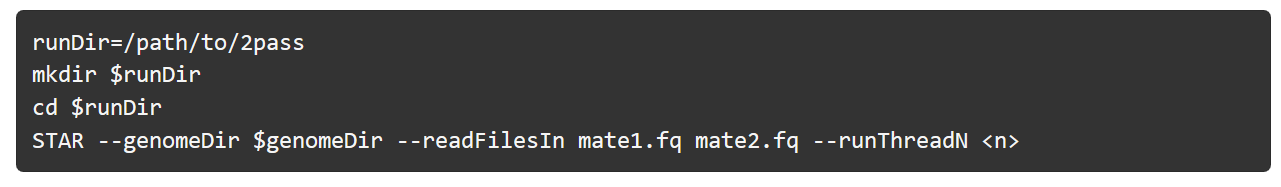
1)raw reads 和 参考基因组比对(推荐使用STAT 2-pass)
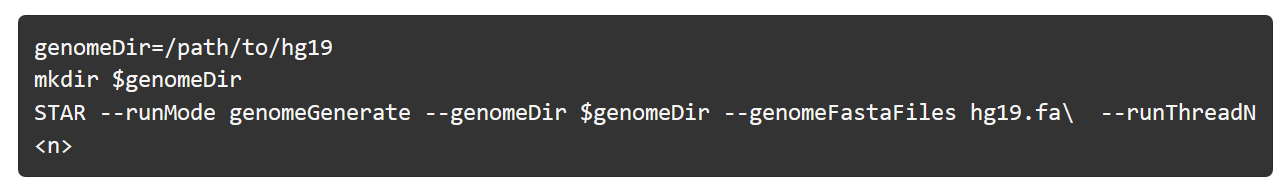
STAR 建立参考基因组的索引
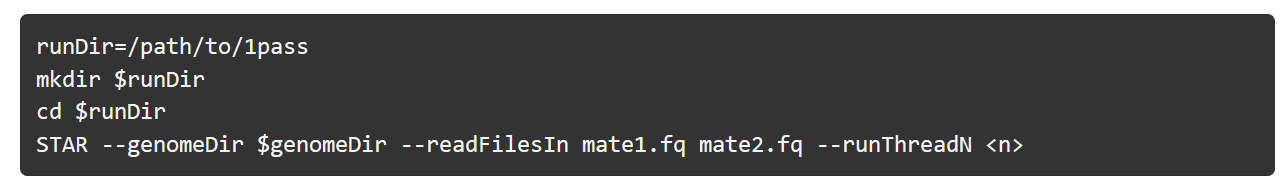
1-pass 比对:
先用第一次比对生成的SJ.out.tab 文件,重新建立索引:
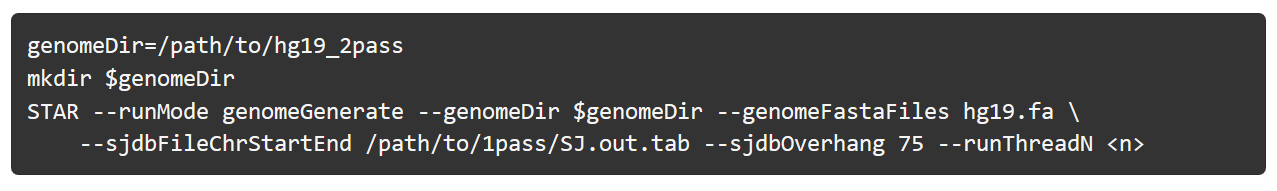
2-pass 比对:
2) mark duplicates and sort
picard 标记重复序列,并sort

3) split N and reassignMappingQuality

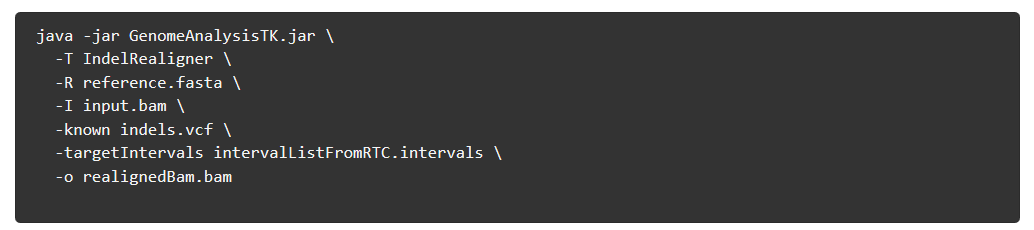
4)Indel realignment (可选的)
5)base recalibration




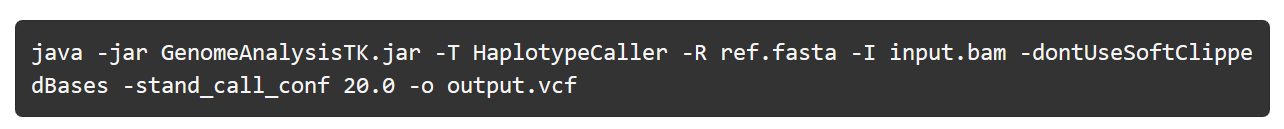
6) variant calling
7)variant filter

参考资料:
https://software.broadinstitute.org/gatk/documentation/article.php?id=3891
https://software.broadinstitute.org/gatk/documentation/article?id=2801