http://blog.sciencenet.cn/blog-2985160-950873.html
利用R/Bioconductor分析结肠癌细胞系差异表达基因
利用R/Bioconductor进行基因差异表达分析。首先从NCBI下载GSE46549数据(GSE46549 中包含了6中不同的结肠癌细胞系,HCT116, HT-29,RKO, SW480, LIM1512 和 CaCo2。链接:http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE46549)。所用的芯片平台是Affymetrix HumanGene 1.0 ST Array 。在R/Bioconductor中, 我们可以用“affy”软件包来读取这些原始数据 。
# 用“affy”软件包来读取这些原始数据
# 用“simpleaffy”软件包中的功能来查看用于做芯片的RNA有没有发生降解
> library (affy)
> library (simpleaffy)
# 用打开浏览器的方式来读取原始的CEL文件
> colon_data <- ReadAffy(widget=TRUE)
# 显示数据信息
> colon_data
AffyBatch object
size of arrays=1050x1050 features (20 kb)
cdf=HuGene-1_0-st-v1 (32321 affyids)
number of samples=12
number of genes=32321
annotation=hugene10stv1
notes=
# 定义12种不同的颜色
> col <- rainbow(12)
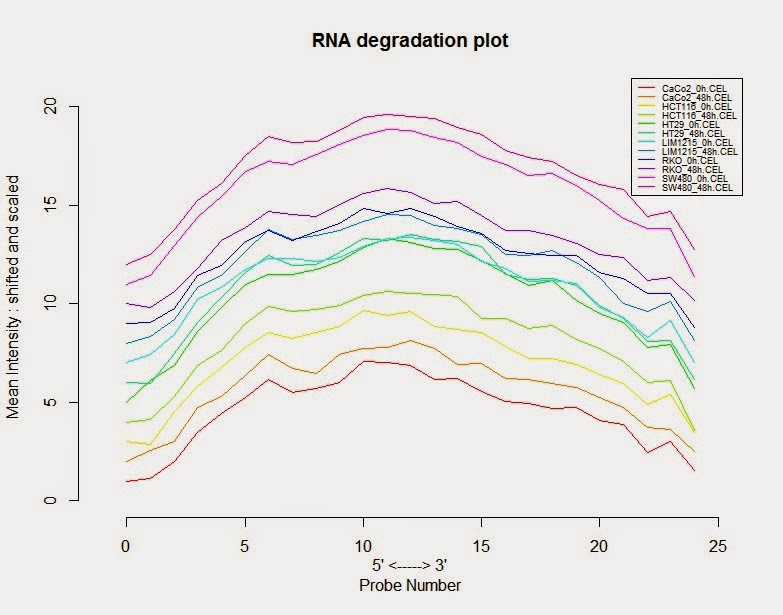
# 评估RNA是不是发生降解
> RNA_deg <- AffyRNAdeg (colon_data)
# 绘制RNA降解图
> plotAffyRNAdeg(RNA_deg, col=col)
>legend(legend=sampleNames(colon_data), x="topright", lty=1, cex=0.55,col=col)
# 查看曲线倾斜度总体是不是相似,有没有个别样品有很大的不同,如果有可能发生降解
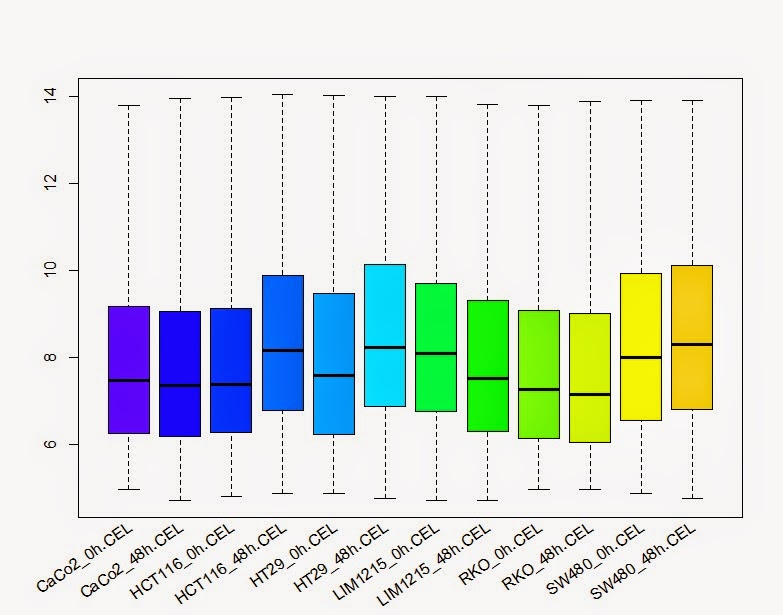
# 箱线图查看每个样品的总体表达
> boxplot(colon_data, col=col)
> lab <- sampleNames (colon_data)
> text(x = seq_along(lab), y =par("usr")[3]-0.2, srt = 35, adj = 1, labels = lab, xpd = TRUE)
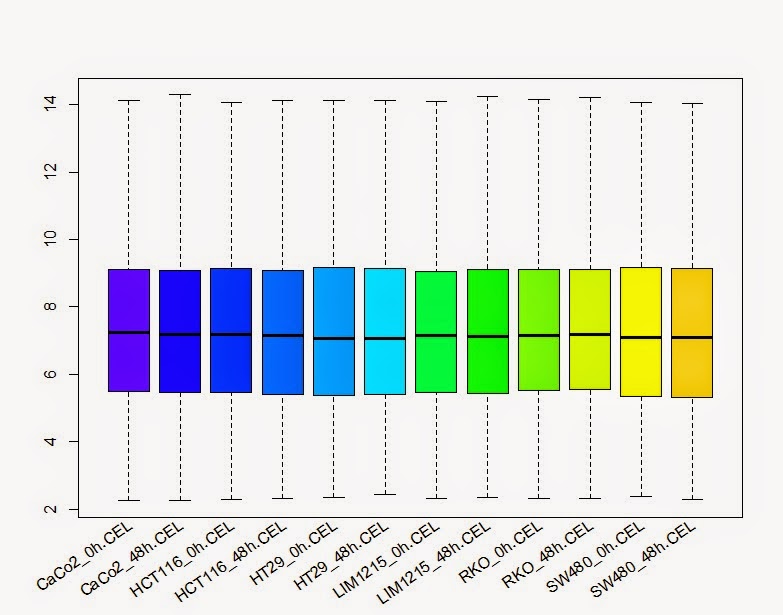
# 用rma 方法对原始的数据进行归一化
> colon_data_normal <-rma(colon_data)
Background correcting
Normalizing
Calculating Expression
#获得基因的表达
> colon_data_exp <-exprs(colon_data_normal)
#箱线图查看归一化后每个样品的总体表达
> boxplot(colon_data_exp,xaxt="n", col=col)
> text(x = seq_along(lab), y =par("usr")[3]-0.2, srt = 35, adj = 1, labels = lab, xpd = TRUE)
#聚类分析
# 计算样品之间的距离
> distance <-dist(t(colon_data_exp))
# 进行聚类并绘制聚类图
> hc <- hclust(distance,method="ave")
> plot(hc, hang=-1)
# 将聚类书分成四簇
> rect.hclust(hc, k=4)
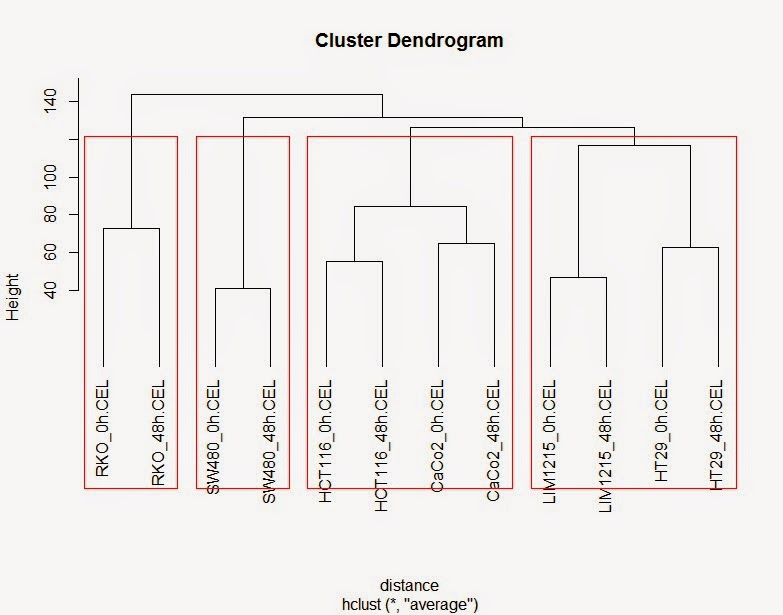
> colon_data_exp <-as.data.frame(colon_data_exp)
> names (colon_data_exp) <-c("CaCo2_0h", "CaCo2_48h", "HCT116_0h","HCT116_48h", "HT29_0h", "HT29_48h","LIM1215_0h", "LIM1215_48h", "RKO_0h","RKO_48h", "SW480_0h", "SW480_48h" )
> head(colon_data_exp)
CaCo2_0h CaCo2_48h HCT116_0hHCT116_48h HT29_0h HT29_48h LIM1215_0h LIM1215_48h RKO_0h RKO_48h SW480_0h SW480_48h
7892501 6.752377 6.592593 7.576409 6.685253 6.006243 6.271173 4.511232 5.958222 5.371307 6.156128 5.779157 5.976742
7892502 6.846297 6.166110 6.173140 5.301924 5.895528 6.652065 5.657238 5.858828 6.478357 6.632395 5.883862 5.483908
7892503 3.592670 4.089133 3.595432 3.983314 3.468884 3.329923 3.598740 4.242956 4.065588 3.548275 3.785297 3.397205
7892504 9.180533 9.308925 8.655324 8.786714 8.101120 8.157576 9.133334 8.804358 8.933208 9.067537 8.494151 8.202562
7892505 3.966066 4.963173 4.181195 4.357158 4.238690 4.334786 3.705270 4.698990 3.530189 4.019916 4.057861 3.633473
7892506 6.161011 5.537104 5.048375 5.354080 6.311650 6.777239 6.325180 6.337767 5.204783 6.191189 6.470301 5.762595
#利用 corrgram软件包中的 corrgram 方法来查看样品之间的相关性
> library (corrgram)
> corrgram(cor(colon_data_exp),lower.panel=panel.conf, upper.panel=panel.pie, col.regions=cm.colors,main="Sample Correlation")
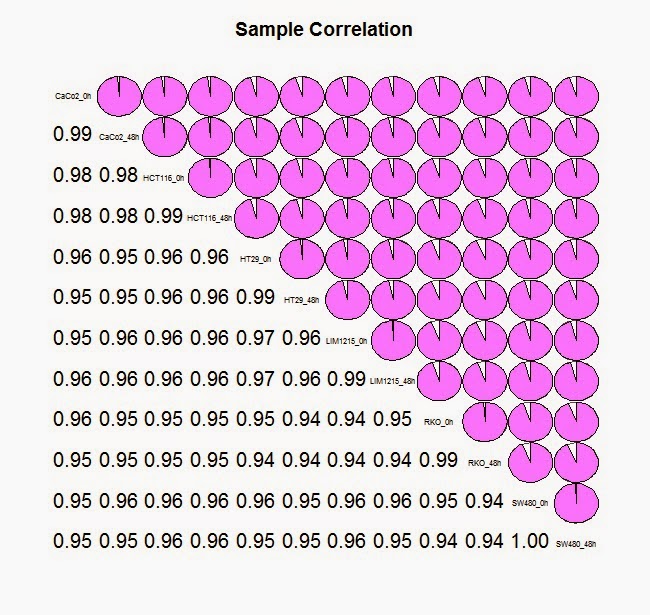
# 加载 limma 软件包, 用线性模型来进行基因差异化分析.
> library (limma)
> groups <- c("CaCo2","CaCo2", "HCT116", "HCT116", "HT29","HT29", "LIM1215", "LIM1215", "RKO","RKO", "SW480", "SW480")
> groups <- as.factor(groups)
> des <- model.matrix(~groups)
> des
(Intercept) groupsHCT116groupsHT29 groupsLIM1215 groupsRKO groupsSW480
1 1 0 0 0 0 0
2 1 0 0 0 0 0
3 1 1 0 0 0 0
4 1 1 0 0 0 0
5 1 0 1 0 0 0
6 1 0 1 0 0 0
7 1 0 0 1 0 0
8 1 0 0 1 0 0
9 1 0 0 0 1 0
10 1 0 0 0 1 0
11 1 0 0 0 0 1
12 1 0 0 0 0 1
attr(,"assign")
[1] 0 1 1 1 1 1
attr(,"contrasts")
attr(,"contrasts")$groups
[1] "contr.treatment"
> fit <- lmFit(colon_data_exp, des)
> fit <- eBayes(fit)
> toptable(fit, coef=2)
> toptable(fit, coef=3)
> toptable(fit, coef=4)
> toptable(fit, coef=5)
> toptable(fit, coef=6)
logFC t P.Value adj.P.Val B
8139212 6.388442 65.600931.119720e-13 3.619048e-09 19.99287
7918694 5.064495 44.597623.955354e-12 4.240144e-08 17.78992
7963427 6.126554 43.938644.537555e-12 4.240144e-08 17.69051
7906079 -5.324317 -42.03273 6.830113e-124.240144e-08 17.38849
7923547 5.250144 41.458347.753910e-12 4.240144e-08 17.29302
8168557 -5.885267 -41.27382 8.079409e-124.240144e-08 17.26189
8166611 -6.039658 -40.70440 9.183197e-124.240144e-08 17.16440
8088180 4.370571 38.742431.447748e-11 4.913646e-08 16.81113
7922337 -5.164845 -38.60976 1.494225e-114.913646e-08 16.78623
7991070 4.059239 38.278071.617830e-11 4.913646e-08 16.72337
# 获取每组前5000 个差异基因
> diff_2 <- toptable(fit, coef=2,number=5000)
> diff_3 <- toptable(fit, coef=3,number=5000)
> diff_4 <- toptable(fit, coef=4,number=5000)
> diff_5 <- toptable(fit, coef=5,number=5000)
> diff_6 <- toptable(fit, coef=6,number=5000)
# 获取1.5倍差异的基因FDR < 0.01
> diff_2 <- diff_2[abs(diff_2$logFC)>= 1.5 & diff_2$adj.P.Val < 0.01,]
> diff_3 <- diff_3[abs(diff_2$logFC)>= 1.5 & diff_3$adj.P.Val < 0.01,]
> diff_3 <- diff_3[abs(diff_3$logFC)>= 1.5 & diff_3$adj.P.Val < 0.01,]
> diff_4 <- diff_4[abs(diff_4$logFC)>= 1.5 & diff_4$adj.P.Val < 0.01,]
> diff_5 <- diff_5[abs(diff_5$logFC)>= 1.5 & diff_5$adj.P.Val < 0.01,]
> diff_6 <- diff_6[abs(diff_6$logFC)>= 1.5 & diff_6$adj.P.Val < 0.01,]
> row_2 <- rownames(diff_2)
> row_3 <- rownames(diff_3)
> row_4 <- rownames(diff_4)
> row_5 <- rownames(diff_5)
> row_6 <- rownames(diff_6)
> diff_row_name <- union(row_2,row_3)
> diff_row_name <-union(diff_row_name, row_4)
> diff_row_name <- union(diff_row_name,row_5)
> diff_row_name <-union(diff_row_name, row_6)
> colon_diff_exp <-colon_data_exp[match(diff_row_name, rownames(colon_data_exp)),]
> nrow(colon_diff_exp)
[1] 3184
> library (gplots)
> heatmap.2(as.matrix(colon_diff_exp), trace="none", col=bluered,density.info="none", labRow=F, srtCol=40)
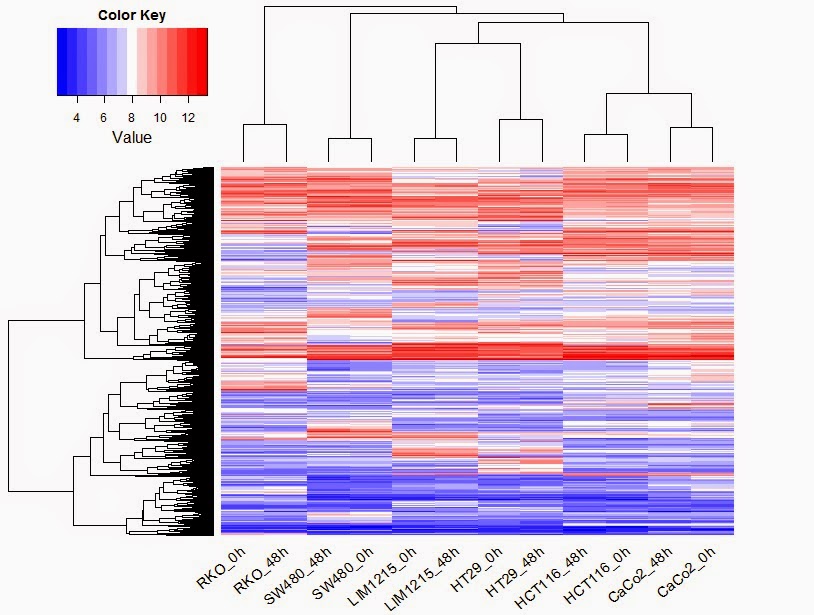
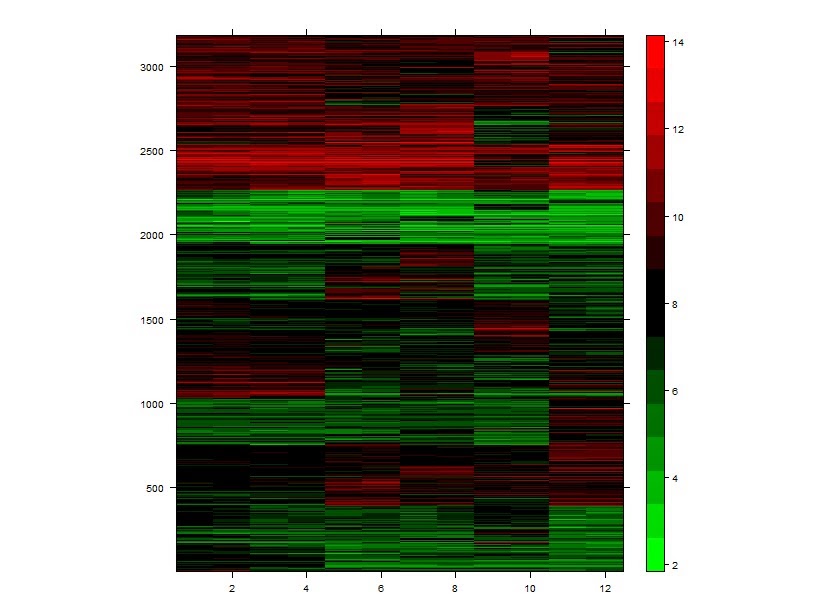
# k-means方法聚类差异基因
> km <- kmeans(colon_diff_exp, 10)
> kmeansclus <- km$cluster
> names(kmeansclus) <- NULL
> km_colon_diff <- cbind(colon_diff_exp, kmeansclus)
> km_colon_diff <-km_colon_diff[order(km_colon_diff$kmeansclus),]
> library (lattice)
> levelplot (as.matrix (t(km_colon_diff)), col.regions=cm.colors,aspect=1.2, xlab="", ylab="")
> RKO<-rowMeans(colon_diff_exp[,9:10])
> CaCo2<-rowMeans(colon_diff_exp[,1:2])
> HCT116 <-rowMeans(colon_diff_exp[,3:4])
> HT29<-rowMeans(colon_diff_exp[,5:6])
> LIM1215<-rowMeans(colon_diff_exp[,7:8])
> SW480<-rowMeans(colon_diff_exp[,11:12])
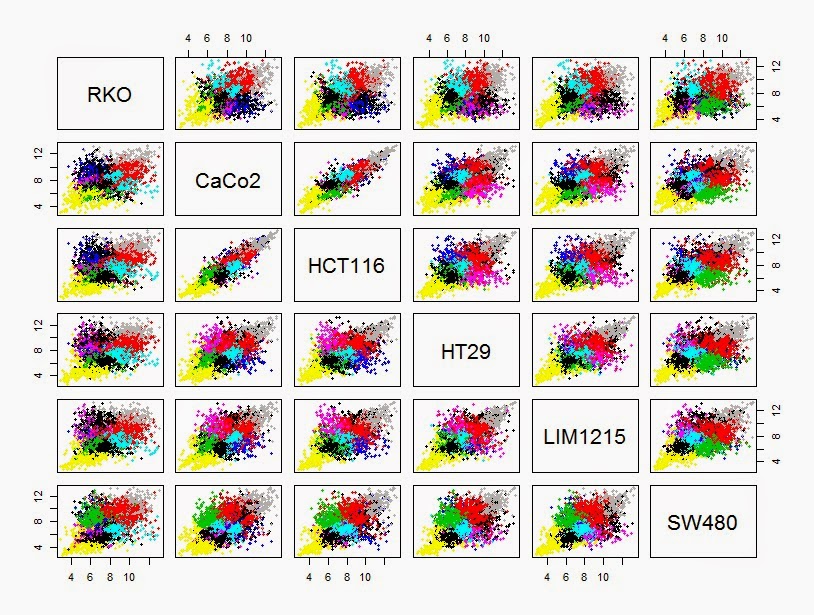
> mean_diff_exp <- cbind(RKO, CaCo2,HCT116, HT29, LIM1215, SW480)
> pairs(mean_diff_exp,col=kmeansclus,pch=19, cex=0.6)