一、流程:
1.定义好每个原子力场类型:Edit Sets(分组)
2.加H(一般是仅与Al-O八面体中的Al相连的O原子)、给H分组
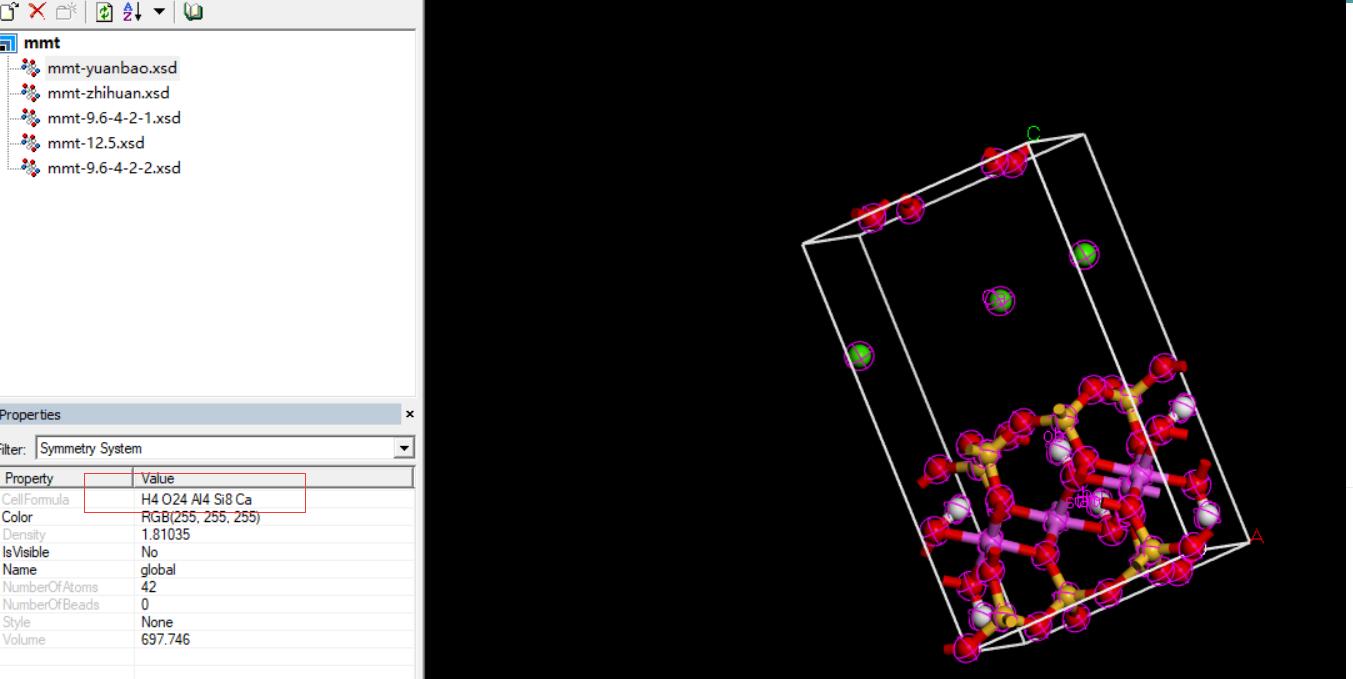
3.“make P1”、校核化学式(正确与否)
4.扩成超晶胞(Na-mmt:4*1*1,Ca-mmt:4*2*1,illite:2*1*1,由化学式而定)
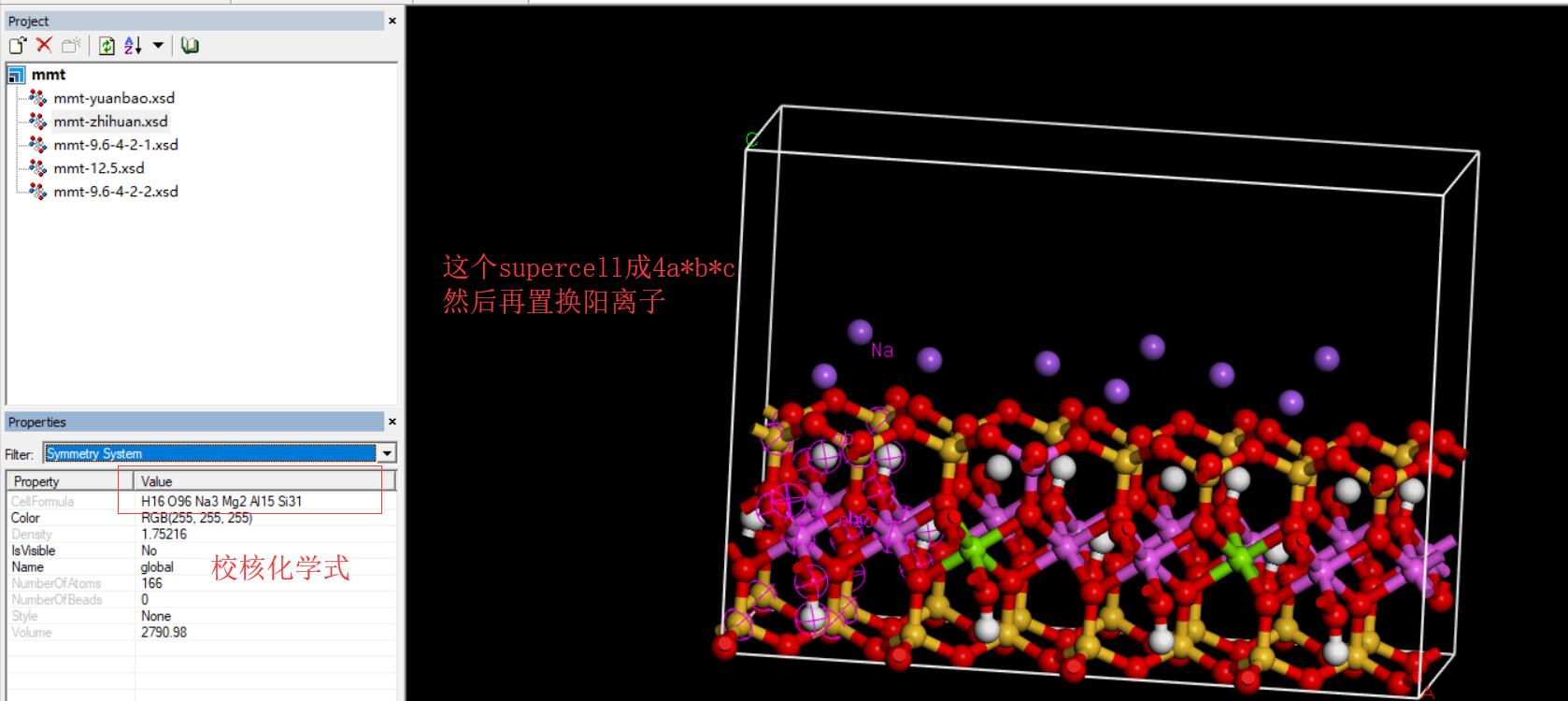
5.置换阳离子:
mmt:模型中每 32 个 Si 4+有 1 个被 Al 3+ 取代,每 8 个 Al 3+ 有 1 个被 Mg 2+ 取代,因此晶格取代产生的层电荷为-0.75e/元胞,由层间
Na + 离 子 平 衡 。 建 立 的 蒙 脱 石 的 分 子 式 为 :Na 0.75 (Si 7.75 Al 0.25 )(Al 3.5 Mg 0.5 )O 20 (OH) 4 。
4a*b*c:Na 3 (Si 31 Al )(Al 14 Mg 2 )O 80 (OH) 16;所以,要用1个Al(at)去取代1个Si(st),注意,这里是四面体中的Al(at),不是八面体中的Al(ao);
以及2个Mg(mgo)去取代八面体中的2个Al(ao)。
Mg置换Al-O八面体的Al,与Mg相连的ob——obos(4个),oh——ohs(2个);
Al置换Si-O四面体的Si,与Al相连的ob——obts(3个)。
层间阳离子:直接改,随机充填在层间域中(包括水分子)
6.调整c值(可以在左下角设置,先选盒子)、扩胞:根据实际需求来调整。
0——0.96nm(0个水分子);
1——1.25(32个水分子);
2——1.53(64个水分子);
3——1.85(96个水分子);
4——2.1(144个水分子)。

7.优化
8.计算
(进入工作目录kao
cd kao/
msi2lmp.exe kao -class I -frc clayff -i -ignore > out.log
生成后kao文件夹会有data生成,可检查一下data力场对不对,不对的话检查一下frc_files里面的clayff.frc参数准不准。)
9.打开putty,输入:
source /opt/software/impi/mpivars.sh (注意:只有source后边是有空格,其他的没有空格)
mpirun -np 4 lmp_mpi < in.elastic(计算文件名,如:mmt422.in)
(4代表4核,也可以选择8,10等)

ssh node02 回车(进入新服务器)
10.输入计算命令:
lmp_mkl < xxx.in > xxx.out (后部分的out文金价可以不生成出来)