如果只有SNP的染色体和物理位置信息,该如何批量转换得到 rs ID?

思路非常简单,只需要下载 dbSNP 的参考文件,根据位置信息从参考文件中获取对应的 rs 编号即可。
下面列举两个例子。
重命名 PLINK 文件 SNP 名字
第一个例子是 PLINK 格式的文件,要把 .bim 文件中的 SNP 名字改为 rs id。
首先从 UCSC 下载纯文本格式的 dbSNP release 151 并解压,这里下载的是 hg19 版本:
wget http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/snp151.txt.gz
gzip snp151.txt.gz -d
snp151.txt.gz 包含了所有 SNPs,总共有 12G 大。如果只需要常见 SNPs,或者说硬盘不够大,则可以下载只有常见 SNPs 的文件,只有 748M:
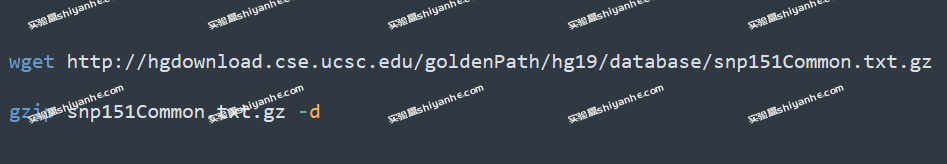
注意,下载的文件的 chromStart 列是 0-based 的。0-based 指的是染色体坐标从 0 开始,第一个位置记为 0,而 1-based 则是从 1 开始算出。如果还是不明白 0-based 是什么意思,可以看看 Chromosome coordinate systems: 0-based, 1-based。
接下来,新建一个 Python 脚本,脚本名字为 rename_snps_shiyanhe.py:
# 欢迎访问实验盒www.shiyanhe.com
import sys
snps = {'snp_%s_%s' % (e[0][3:], e[1]): e[2] for e in (l.strip().split() for l in open(sys.argv[1]))}
bim = (l.strip().split() for l in open(sys.argv[2]))
new = open(sys.argv[3], 'w')
for e in bim:
e[1] = snps.get(e[1], e[1])
new.write(' '.join(e) + '
')
new.close()
bim.close()
按 rename_snps_shiyanhe.py SNP参考文件 原来的bim 新的bim文件 这样的命令执行脚本,比如:
python rename_snps_shiyanhe.py snp151.txt original.bim new.bim
根据 VCF 文件查询 rs id
如果要查询 vcf 文件的 SNPs,根据前面下载的参考文件,自己写脚本即可。不过,如果不会写脚本,也可以用下面介绍的用 BEDOPS 的方法。
根据基因组坐标版本下载 dbSNP 数据,这里下载 hg38 版本:
vcf = ftp://ftp.ncbi.nih.gov/snp/organisms/human_9606_b151_GRCh38p7/VCF/All_20180418.vcf.gz
wget -qO- ${VCF}
| gunzip -c -
| convert2bed --input=vcf --sort-tmpdir=${PWD} -
| awk '{ print "chr"$0; }' -
| starch --omit-signature -
> All_20180418.starch
如果硬盘空间不够,也可下载常见 SNPs 的参考文件,路径为 ftp://ftp.ncbi.nih.gov/snp/organisms/human_9606_b151_GRCh38p7/VCF/common_all_20180418.vcf.gz 。
假设要重命名的 vcf 文件为 shiyanhe.com.vcf ,可以这么查询:
bedops --element-of 1 All_20180418.starch <(vcf2bed < shiyanhe.com.vcf) | cut -f4 > rsid.txt
如果想得到 bed 文件:
bedops --element-of 1 all_20180418.starch <(vcf2bed < shiyanhe.com.vcf) > shiyanhe.com.recoded.bed
参考
