samtools flagstat命令简介:
统计输入文件的相关数据并将这些数据输出至屏幕显示。每一项统计数据都由两部分组成,分别是QC pass和QC failed,表示通过QC的reads数据量和未通过QC的reads数量。以“PASS + FAILED”格式显示。还可以根据samtools的标志位显示相应的内容,但是这里不做讨论。
命令格式:
samtools flagstat <in.bam> |<in.sam> | <in.cram>
运行flagstat命令得到的结果如下图所示。
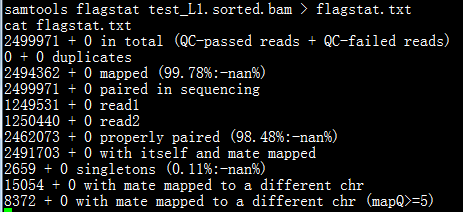
从第一行至第十一行分别表示:
1. QC pass的reads的数量为2499917,未通过QC的reads数量为0,意味着一共有2499971条reads;
2. 重复reads的数量,QC pass和failed
3. 比对到参考基因组上的reads数量;
4. paired reads数据数量;
5. read1的数量;
6. read2 的数量;
7. 正确地匹配到参考序列的reads数量;
8. 一对reads都比对到了参考序列上的数量,但是并不一定比对到同一条染色体上;
9. 一对reads中只有一条与参考序列相匹配的数量;
10. 一对reads比对到不同染色体的数量;
11. 一对reads比对到不同染色体的且比对质量值大于5的数量。