本节课程,需要完成扩增子分析解读1质控 实验设计 双端序列合并和2提取barcode 质控及样品拆分 切除扩增引物
先看一下扩增子分析的整体流程,从下向上逐层分析
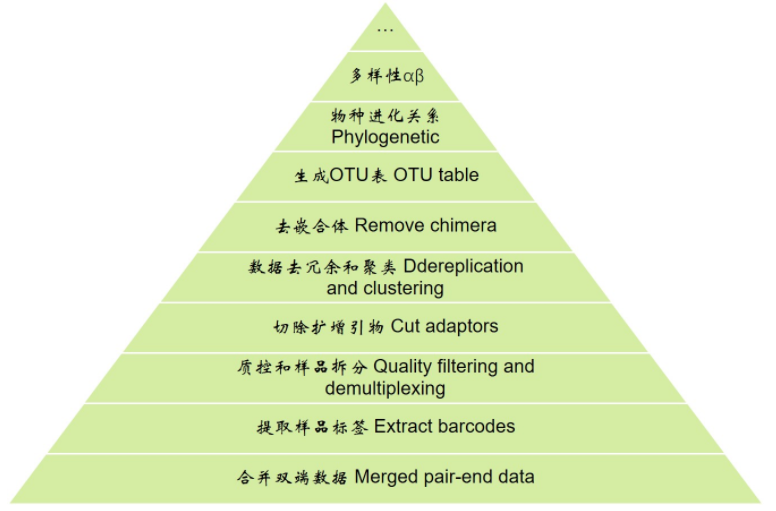
分析前准备
# 进入工作目录 cd example_PE250
上一节回顾:我们提取barcode,质控及样品拆分,切除扩增引物,经历了两节课6步数据处理才拿到我们扩增的高质量目的片段(貌似基因组/RNA-Seq测序结果直接就是这个阶段了,可以直接mapping)
接下来我们将这些序列去冗余、聚类为OTU、再去除嵌合体,这样就可以获得高质量的OTU(类似于参考基因组/转录组),用于定量分析每个OTU的丰度。这一阶段我们使用著名的扩增子分析流程Usearch。
Usearch简介
http://drive5.com/usearch/
Usearch之前介绍过http://www.cnblogs.com/freescience/p/7270861.html
软件作者不仅有Usearch一款软件,它的Muscle(多序列比对,引用18659+4212次),Uparse(OTU聚类算法,引用1529次), Uchime(扩增子嵌合体检测,引用3558次)等众多流行工具,个人引用超4万次,而且发的软件大多由作者一人完成,佩服。
Usearch安装
这个软件64位版收费,但32位对任何人免费,可以在下面网址下载 http://www.drive5.com/usearch/download.html 同意许可协议,选择软件版本(5.2 — 10.0),选择运行平台(Linux, Windows或Mac OSX)填写邮箱获得下载地址。不允许私人传播。
这里我选择10.0版本,系统选择Linux。
收到的邮件中第一个链接即下载地址,后面两个链接为帮助文档和安装说明,先不用管,按我下面的操作来。
# 下载程序并重命名:下载链接来自邮件,请用户自行复制邮件中地址替换下面代码中的网址;或者在windows里面下载并重命名为usearch10 wget -O "usearch10" http://drive5.com/cgi-bin/upload3.py?license=XXXXXX # 添加可执行权限 chmod +x usearch10 # 运行程序测试,成功可显示程序版本、系统信息和用户授权信息 ./usearch10
7. 格式转换
做生信为什么要学Python/Perl/Shell这些语言,主要原因是各软件间要求的具体格式不同,需要进行格式转换,才能继续运行。因此想成为高手,不会语言基本寸步难行。
我们现在将QIIME拆分的结果类型,要转换成Usearch要求的格式。常见的解决思路是读Usearch帮助看它的格式要求,写个Python/Perl脚本转换格式。我这里使用了Shell脚本一行解决,优点是快,但缺点很多(人不容易看懂、不同Linux系统shell版本不同可能失效)
我们要转换的序列文件其实一直是fasta格式,只是序列名称行格式不同
# 目前格式>KO1_0 HISEQ:419:H55JGBCXY:1:1101:1931:2086 1:N:0:CACGAT orig_bc=TAGCTT new_bc=TAGCTT bc_diffs=0# Usearch要求的格式>KO1_0;barcodelabel=KO1;
# 格式转换 sed 's/ .*/;/g;s/>.*/&&/g;s/;>/;barcodelabel=/g;s/_[0-9]*;$/;/g' temp/PE250_P5.fa > temp/seqs_usearch.fa
上面这条命令有点复杂。sed是linux的一条命令,又是一种语言,擅长文本替换。替换的思路分四步:首先s/ ./;/g将原文件空格后面的内容(全是无用信息)替换为分号;其次s/>./&&/g是将序列名重复一次;再次s/;>/;barcodelabel=/g将重复后的;>替换为;barcodelabel=;最后s/_[0-9]*;$/;/g替换序列编号为分号。这只是我的思路,分析数据如解答数学题,可以有多种解法,你够聪明还会想出更好的解法。
新人一定感觉这命令每句都不像人话,我告诉你Perl和Shell就是这样—难读但高效。改用易读的Python语言,肯定没有Shell简洁。
8. 去冗余
为什么要去冗余?
因为原始序列几百万条,聚类计算的时间极其恐怖。而已知扩增子测序结果中序列重复度高,并且大量出现1次或几次的序列统计学和功能上意义不大。因此将几百万条序列去冗余,并过滤低丰度序列,一般只剩几万条,极大的减少了下游分析的工作量,并可使结果更容易理解。
usearch10的去冗余命令叫-fastx_uniques,紧跟着输入文件;
-fastaout 接输出文件;
-minuniquesize 参数设置保留的最小丰度reads数,建议最小设置为2,去掉所有的单次出现序列(singletons),数据量大建议设置总数据量的百万分之一并取整数部分
-sizeout 在序列名称中添加序列出现的频率
# 序列去冗余 ./usearch10 -fastx_uniques temp/seqs_usearch.fa -fastaout temp/seqs_unique.fa -minuniquesize 2 -sizeout
计算过程中出现如下信息:
00:06 607Mb 100.0% Reading temp/seqs_usearch.fa00:06 574Mb CPU has 96 cores, defaulting to 10 threads00:08 915Mb 100.0% DF00:09 935Mb 1268345 seqs, 686530 uniques, 624363 singletons (90.9%)00:09 935Mb Min size 1, median 1, max 18774, avg 1.8562167 uniques written, 182874 clusters size < 2 discarded (26.6%)
主要内容为读取输入文件;
检查到系统有96个CPU,默认使用了10个线程;
总共有1268345条序列,其中非重复的序列有686530个,非重复且只出现一次的有624363个(90.9%的非冗余序列是singletons,多吗?);
最小值、中位数、最大值、平均值;输出结果有62167个结果,丢弃掉的数据占26.6%。
本条命令的详细使用,请阅读官方文档 http://www.drive5.com/usearch/manual/cmd_fastx_uniques.html
9. 聚类OTU
为什么要聚类OTU?
是因为Unique的序列仍然远多于物种数量,并且扩增的物种可能存在rDNA的多拷贝且存在变异而得到来自同一物种的多条序列扩增结果。目前人为定义序列相似度通常97%以上为OTU,大约是物种分类学种的水平,实际上1个OTU可能包括多个物种,而一个物种也可能扩增出多个OTU。
下面我们用usearch10将非冗余的序列聚类
-cluster_otus接输入文件;
-otus后面为输出的otu文件的fasta格式;
-uparseout输出聚类的具体细节
-relabel Otu为重命名序列以Otu起始
# 聚类OTU ./usearch10 -cluster_otus temp/seqs_unique.fa -otus temp/otus.fa -uparseout temp/uparse.txt -relabel Otu
程序运行过程会显示运行时间、进度,发现的OTU,以及嵌合体数据;结果如下:
04:11 84Mb 100.0% 5489 OTUs, 9209 chimeras
程序一共运行了3分39秒,聚类发现5486个OTUs,同时发现了9187个嵌合体并已被丢弃。
Usearch聚类算法之所以能发表在Nature Method上,就是因为其算法UParse在非常强的嵌合体检测能力,对人工重组数据评估,更接近真实结果。下一节我们将详细讲嵌合体产生的原因,以及去除的原理。
本条命令的详细使用,请阅读官方文档 http://www.drive5.com/usearch/manual/cmd_cluster_otus.html
小技巧:统计fasta文件中序列的数量
fasta文件每条序列以大于号(>)开始,其数量与序列数量相同,使用grep检索含有>的行,同时用-c参数对数量进行统计,即可快速获得fasta文件中序列数量。
# 查看OTU数量 grep '>' -c temp/otus.fa