1) Introduction
为了理解基因组数据,通常旨在在基因组浏览器中绘制这样的数据,以及各种基因组注释特征,例如基因或转录物模型,CpG岛,重复区域等。这些功能可以从ENSEMBL或UCSC等公共数据库中提取,也可以在内部生成或策划。许多当前可用的基因组浏览器在显示基因组注释数据方面做了合理的工作,并且存在从R内部连接到其中一些的选项(例如,使用rtracklayer包)。然而,这些解决方案都没有提供完整的Rgraphics系统以多种不同方式显示大数值数据的灵活性。 Gviz软件包旨在通过提供结构化可视化框架来绘制基因组坐标上的任何类型的数据来缩小这一差距。它松散地基于GenomeGraphs包,但为了提高性能和灵活性,重新构建了完整的类层次结构以及所有绘图方法,所有绘图都是使用网格图形系统完成的,并且几个专门的注释类允许集成来自UCSC或ENSEMBL等来源的公开可用的基因组注释数据。
2)Basic Features
if("Gviz" %in% rownames(installed.packages()) == FALSE) {source("http://bioconductor.org/biocLite.R");biocLite("Gviz")}
suppressMessages(library(Gviz))
ls('package:Gviz')
library(GenomicRanges)
data(cpgIslands) #以GC岛数据集为例
class(cpgIslands)
chr <- as.character(unique(seqnames(cpgIslands)))
gen <- genome(cpgIslands)
atrack <- AnnotationTrack(cpgIslands, name = "CpG")
plotTracks(atrack)
![]()
gtrack <- GenomeAxisTrack() plotTracks(list(gtrack, atrack))

itrack <- IdeogramTrack(genome = gen, chromosome = chr) plotTracks(list(itrack, gtrack, atrack))
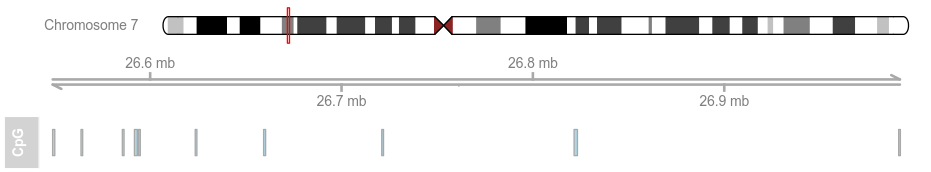
由基本特征可以知道,GVIZ和ggplot2 好像 道理是一样的,一层累积一层添加元素。我们再以下面数据集为例:
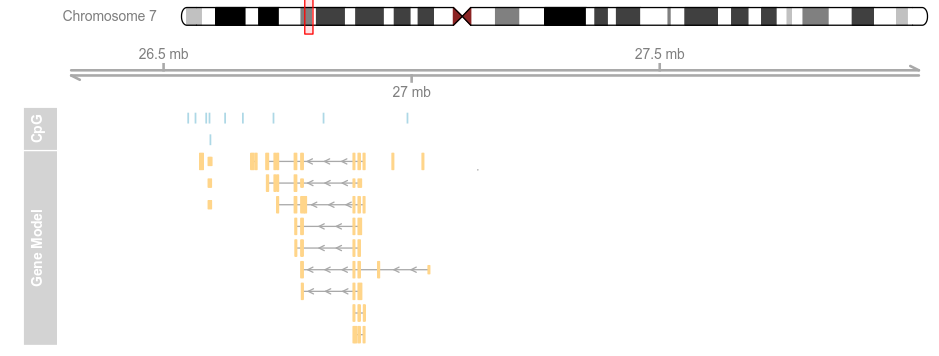
data(geneModels) grtrack <- GeneRegionTrack(geneModels, genome = gen,chromosome = chr, name = "Gene Model") plotTracks(list(itrack, gtrack, atrack, grtrack))
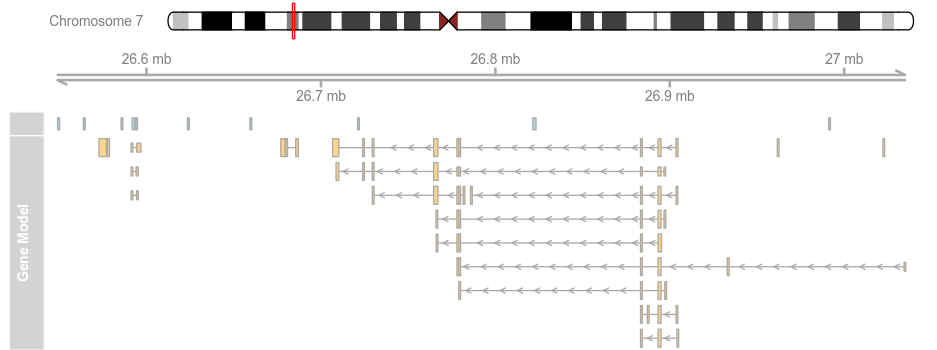
plotTracks(list(itrack, gtrack, atrack, grtrack),from = 26700000, to = 26750000)

plotTracks(list(itrack, gtrack, atrack, grtrack),extend.left = 0.5, extend.right = 1e+06)
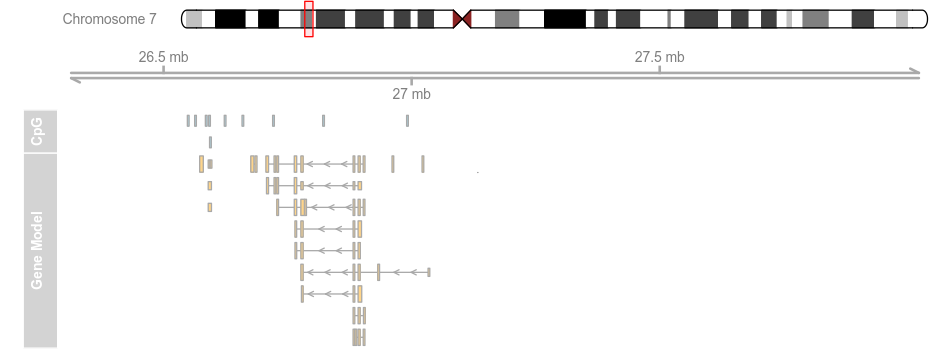
plotTracks(list(itrack, gtrack, atrack, grtrack),
extend.left = 0.5, extend.right = 1e+06, col = NULL)
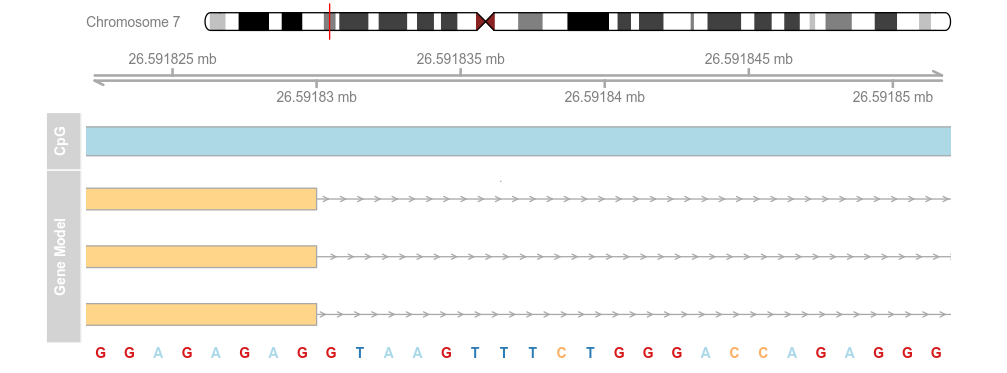
library(BSgenome.Hsapiens.UCSC.hg19)
strack <- SequenceTrack(Hsapiens, chromosome = chr)
plotTracks(list(itrack, gtrack, atrack, grtrack,
strack), from = 26591822, to = 26591852, cex = 0.8)
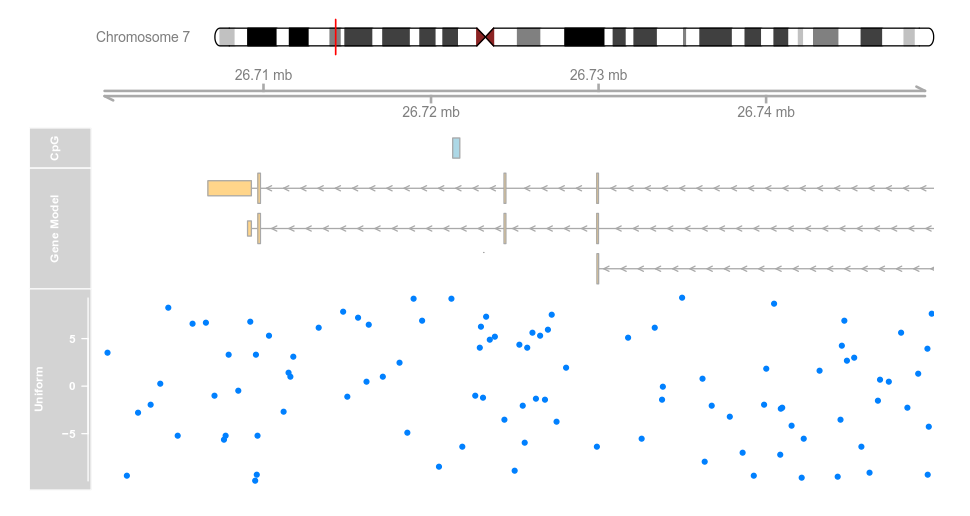
set.seed(255)
lim <- c(26700000, 26750000)
coords <- sort(c(lim[1], sample(seq(from = lim[1],to = lim[2]), 99), lim[2]))
dat <- runif(100, min = -10, max = 10)
dtrack <- DataTrack(data = dat, start = coords[-length(coords)],
end = coords[-1], chromosome = chr, genome = gen,
name = "Uniform")
plotTracks(list(itrack, gtrack, atrack, grtrack,
dtrack), from = lim[1], to = lim[2])
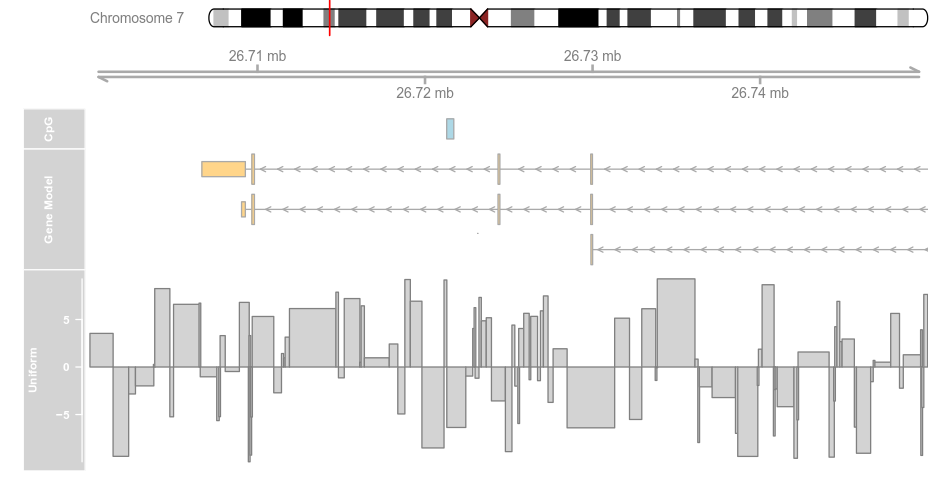
plotTracks(list(itrack, gtrack, atrack, grtrack,
dtrack), from = lim[1], to = lim[2], type = "histogram")
3)Plotting parameters(画图参数调整)
3.1 设置参数
grtrack <- GeneRegionTrack(geneModels, genome = gen,
chromosome = chr, name = "Gene Model", transcriptAnnotation = "symbol",
background.title = "brown")
head(displayPars(grtrack))
plotTracks(list(itrack, gtrack, atrack, grtrack))
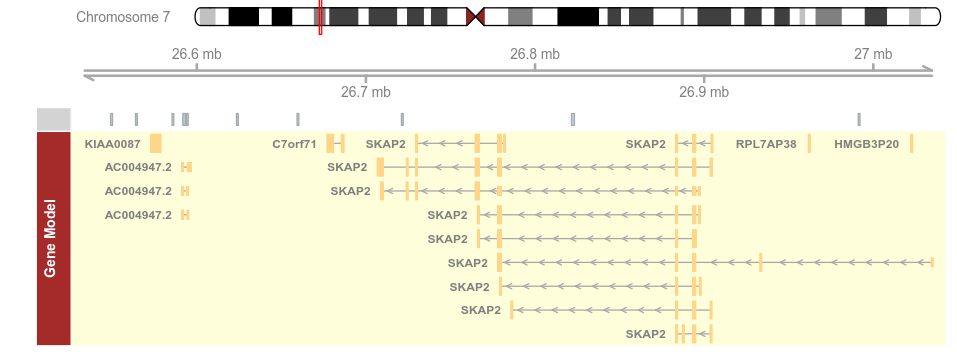
plotTracks(list(itrack, gtrack, atrack, grtrack),
background.panel = "#FFFEDB", background.title = "darkblue")
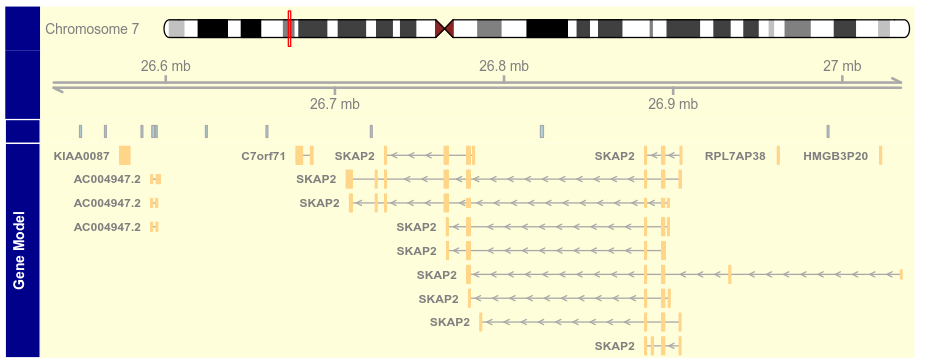
3.2 Schemes(主题)
getOption("Gviz.scheme")
scheme <- getScheme()
scheme$GeneRegionTrack$fill <- "salmon"
scheme$GeneRegionTrack$col <- NULL
scheme$GeneRegionTrack$transcriptAnnotation <- "transcript"
addScheme(scheme, "myScheme")
options(Gviz.scheme = "myScheme")
grtrack <- GeneRegionTrack(geneModels, genome = gen,
chromosome = chr, name = "Gene Model")
plotTracks(grtrack)
options(Gviz.scheme = "default")
grtrack <- GeneRegionTrack(geneModels, genome = gen,
chromosome = chr, name = "Gene Model", transcriptAnnotation = "symbol")
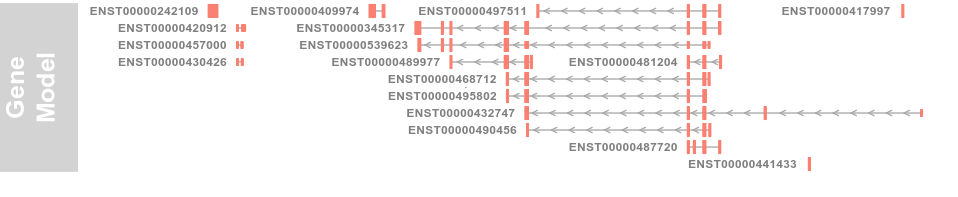
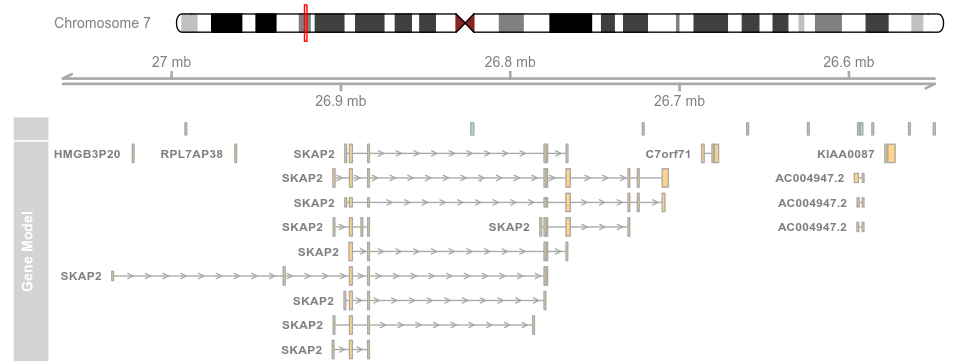
3.3 Plotting direction (方向)
plotTracks(list(itrack, gtrack, atrack, grtrack),reverseStrand = TRUE)
4) Track classes
4.1 GenomeAxisTrack
axisTrack <- GenomeAxisTrack() plotTracks(axisTrack, from = 1e+06, to = 9e+06)

axisTrack <- GenomeAxisTrack(range = IRanges(start = c(2e+06, 4e+06), end = c(3e+06, 7e+06), names = rep("N-stretch",+ 2)))
plotTracks(axisTrack, from = 1e+06, to = 9e+06)
![]()
plotTracks(axisTrack, from = 1e+06, to = 9e+06, showId = TRUE)

plotTracks(axisTrack, from = 1e+06, to = 9e+06, add53 = TRUE,add35 = TRUE)
![]()
plotTracks(axisTrack, from = 1e+06, to = 9e+06, add53 = TRUE,
add35 = TRUE, littleTicks = TRUE)
![]()
plotTracks(axisTrack, from = 1e+06, to = 9e+06, exponent = 4)
![]()
plotTracks(axisTrack, from = 1e+06, to = 9e+06, labelPos = "below")
![]()
plotTracks(axisTrack, from = 1e+06, to = 9e+06, scale = 0.5)
![]()
plotTracks(axisTrack, from = 1e+06, to = 9e+06, scale = 0.5,
labelPos = "below")
4.2) IdeogramTrack
ideoTrack <- IdeogramTrack(genome = "hg19", chromosome = "chrX") plotTracks(ideoTrack, from = 8.5e+07, to = 1.29e+08)
![]()
plotTracks(ideoTrack, from = 8.5e+07, to = 1.29e+08,showId = FALSE)
![]()
plotTracks(ideoTrack, from = 8.5e+07, to = 1.29e+08,
showId = FALSE, showBandId = TRUE, cex.bands = 0.5)
![]()
4.3 DataTrack
data(twoGroups) dTrack <- DataTrack(twoGroups, name = "uniform") plotTracks(dTrack)

plotTracks(dTrack, type = c("boxplot", "a", "g"))
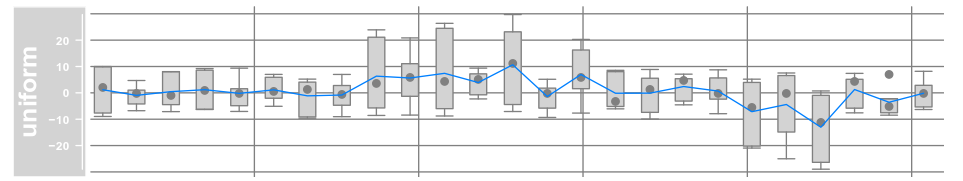
总之,选择不同的画图参数,生成不同的图形
Data Grouping(对于分组数据)
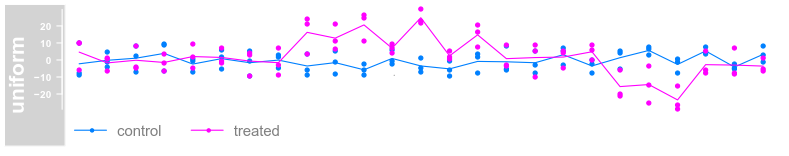
plotTracks(dTrack, groups = rep(c("control", "treated"),
each = 3), type = c("a", "p", "confint"))
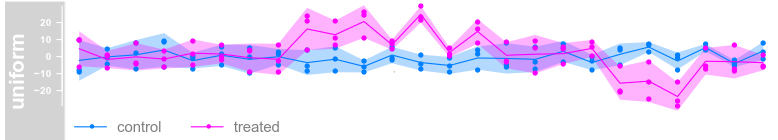
plotTracks(dTrack, groups = rep(c("control", "treated"),
each = 3), type = c("a", "p"), legend = TRUE)
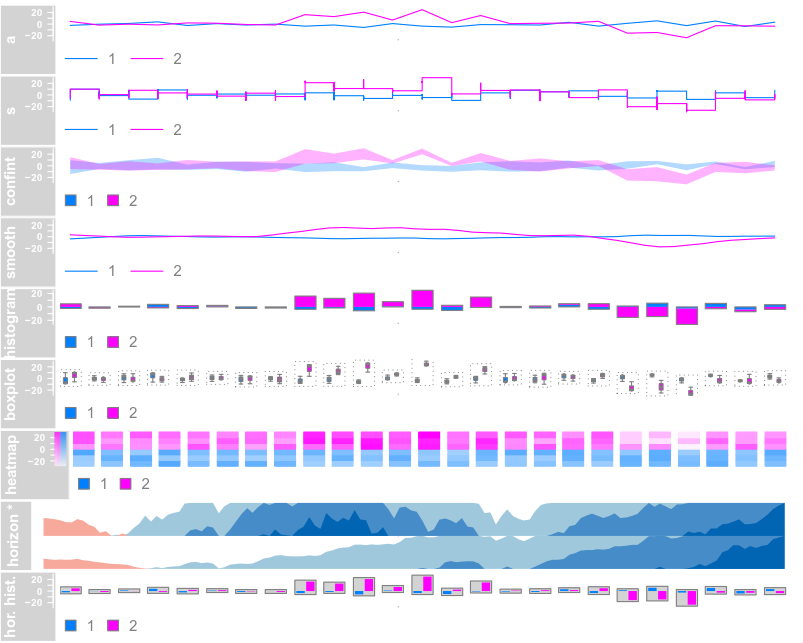
data(dtHoriz)
dtHoriz <- dtHoriz[1:6, ]
plotTracks(dtHoriz, type = "horiz", groups = rownames(values(dtHoriz)),
showSampleNames = TRUE, cex.sampleNames = 0.6,
separator = 1)

总之,选择不同的图形参数类型,生成不同的图形类别
从数据文件中构建 DataTrack objects
bgFile <- system.file("extdata/test.bedGraph", package = "Gviz")
dTrack2 <- DataTrack(range = bgFile, genome = "hg19",
type = "l", chromosome = "chr19", name = "bedGraph")
plotTracks(dTrack2)

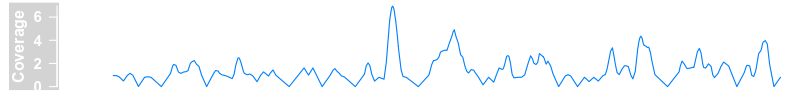
bamFile <- system.file("extdata/test.bam", package = "Gviz")
dTrack4 <- DataTrack(range = bamFile, genome = "hg19", type = "l", name = "Coverage", window = -1, chromosome = "chr1")
plotTracks(dTrack4, from = 189990000, to = 1.9e+08)
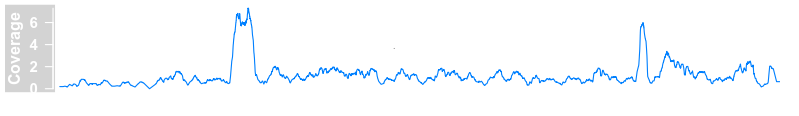
plotTracks(dTrack4, chromosome = "chr1", from = 189891483,to = 190087517)
Data transformations
dat <- sin(seq(pi, 10 * pi, len = 500))
dTrack.big <- DataTrack(start = seq(1, 1e+05, len = 500),
width = 15, chromosome = "chrX", genome = "hg19",
name = "sinus", data = sin(seq(pi, 5 * pi, len = 500)) *
runif(500, 0.5, 1.5))
plotTracks(dTrack.big, type = "hist")
plotTracks(dTrack.big, type = "hist", window = 50)
plotTracks(dTrack.big, type = "hist", window = -1,windowSize = 2500)
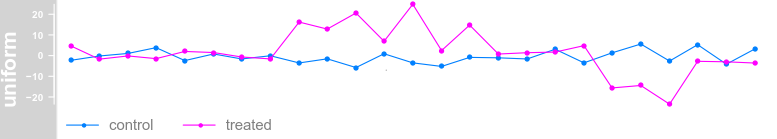
plotTracks(dTrack, groups = rep(c("control", "treated"),
each = 3), type = c("b"), aggregateGroups = TRUE)
4.4 AnnotationTrack
aTrack <- AnnotationTrack(start = c(10, 40, 120),
width = 15, chromosome = "chrX", strand = c("+","*", "-"),
id = c("Huey", "Dewey", "Louie"),
genome = "hg19", name = "foo")
plotTracks(aTrack)
![]()
plotTracks(aTrack, shape = "box", featureAnnotation = "id")
![]()
plotTracks(aTrack, shape = "ellipse", featureAnnotation = "id", fontcolor.feature = "darkblue")
![]()
aTrack.groups <- AnnotationTrack(start = c(50, 180,260, 460, 860, 1240),
width = c(15, 20, 40, 100,00, 20),
chromosome = "chrX",
strand = rep(c("+", "*", "-"), c(1, 3, 2)),
group = rep(c("Huey", "Dewey", "Louie"), c(1, 3, 2)),
genome = "hg19",
name = "foo") + "Dewey", "Louie"), c(1, 3, 2)), genome = "hg19",name = "foo")
plotTracks(aTrack.groups, groupAnnotation = "group")

plotTracks(aTrack.groups, groupAnnotation = "group",just.group = "above")
![]()
aTrack.stacked <- AnnotationTrack(start = c(50, 180,260, 800, 600, 1240), width = c(15, 20, 40, 100,
500, 20), chromosome = "chrX", strand = "*",
group = rep(c("Huey", "Dewey", "Louie"), c(1, 3, 2)),
genome = "hg19", name = "foo")
plotTracks(aTrack.stacked, groupAnnotation = "group")
![]()
plotTracks(aTrack.stacked, stacking = "dense")
![]()
data("denseAnnTrack")
plotTracks(denseAnnTrack, showOverplotting = TRUE)

4.5,4.6,4.7,4.8,4.9回来再写
4.9 AlignmentsTrack
afrom <- 2960000 ato <- 3160000 alTrack <- AlignmentsTrack(system.file(package = "Gviz", "extdata", "gapped.bam"), isPaired = TRUE) bmt <- BiomartGeneRegionTrack(genome = "hg19", chromosome = "chr12",start = afrom, end = ato, filter = list(with_ox_refseq_mrna = TRUE),stacking = "dense") plotTracks(c(bmt, alTrack), from = afrom, to = ato, chromosome = "chr12")
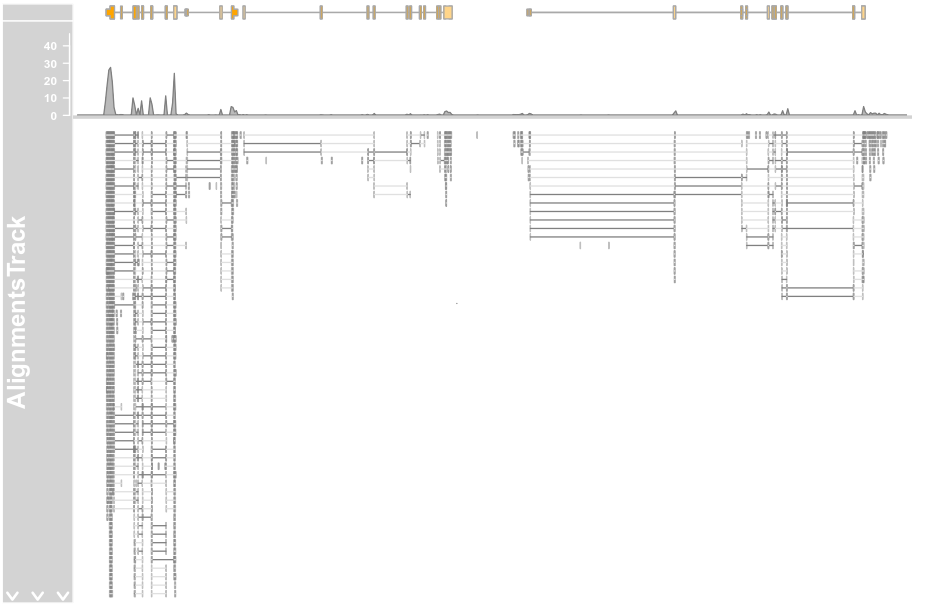
plotTracks(c(bmt, alTrack), from = afrom, to = ato,
chromosome = "chr12", min.height = 0, coverageHeight = 0.08,
minCoverageHeight = 0)
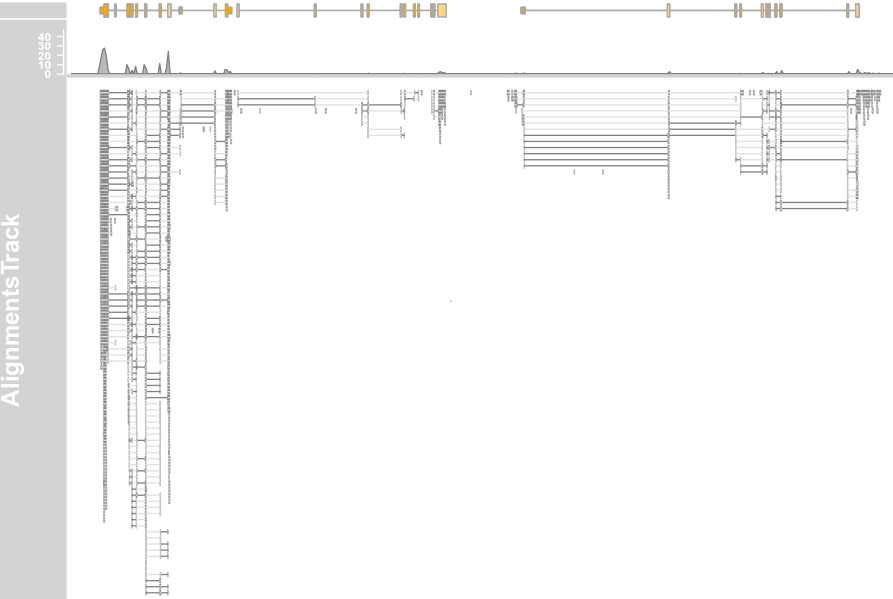
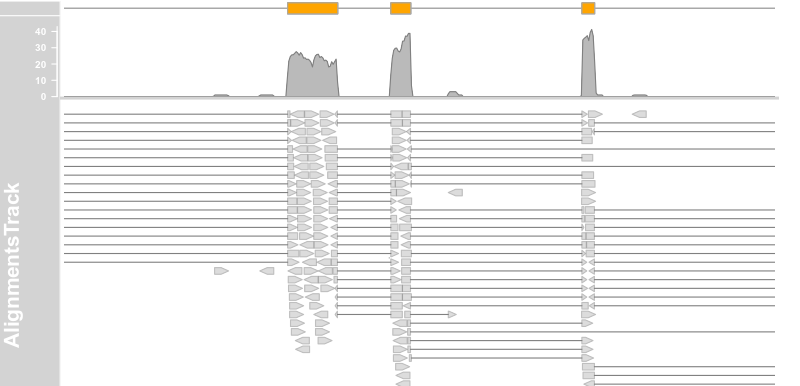
plotTracks(c(alTrack, bmt), from = afrom, to = ato, chromosome = "chr12", type = "coverage")

plotTracks(c(bmt, alTrack), from = afrom + 12700,to = afrom + 15200, chromosome = "chr12")

plotTracks(c(bmt, alTrack), from = afrom + 12700,
to = afrom + 15200, chromosome = "chr12", type = c("coverage","sashimi"))
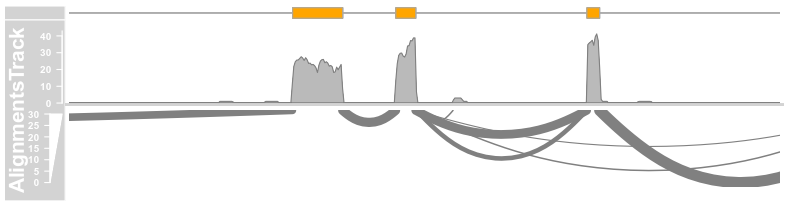
introns <- GRanges("chr12", IRanges(start = c(2973662,2973919), end = c(2973848, 2974520)))
plotTracks(c(bmt, alTrack), from = afrom + 12700,
to = afrom + 15200, chromosome = "chr12", type = c("coverage","sashimi"), sashimiFilter = introns)
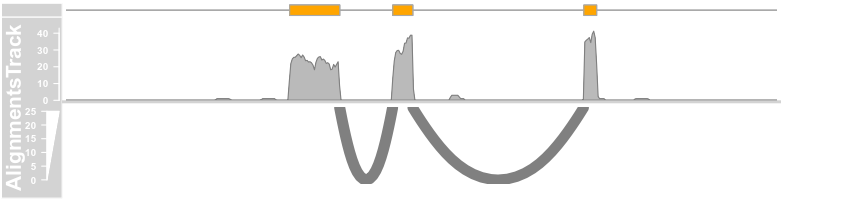
plotTracks(c(bmt, alTrack), from = afrom + 12700,to = afrom + 15200, chromosome = "chr12",
type = c("coverage", "sashimi"), sashimiFilter = introns, sashimiFilterTolerance = 5L)
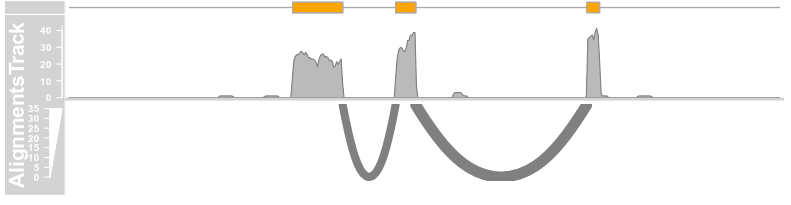
plotTracks(c(bmt, alTrack), from = afrom + 12700,
to = afrom + 15200, chromosome = "chr12", reverseStacking = TRUE,
col.mates = "purple", col.gap = "orange", type = "pileup")
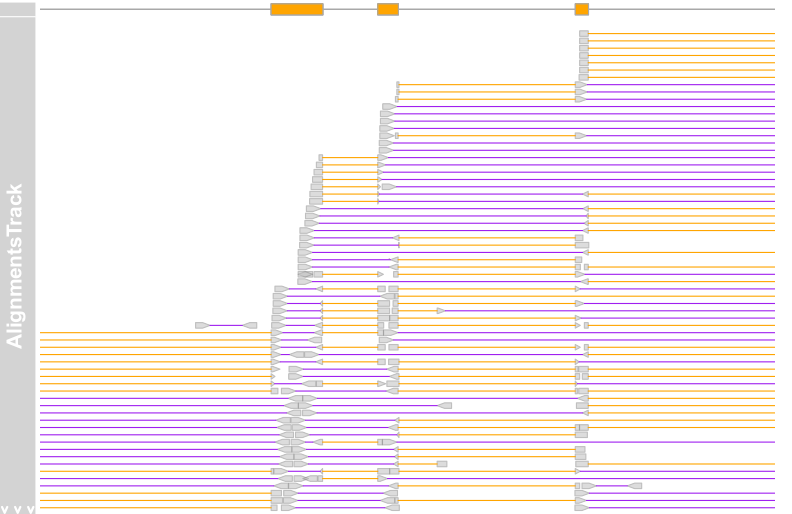
alTrack <- AlignmentsTrack(system.file(package = "Gviz","extdata", "gapped.bam"),isPaired = FALSE) plotTracks(c(bmt, alTrack), from = afrom + 12700,to = afrom + 15200, chromosome = "chr12")
4.10 Creating tracks from UCSC data
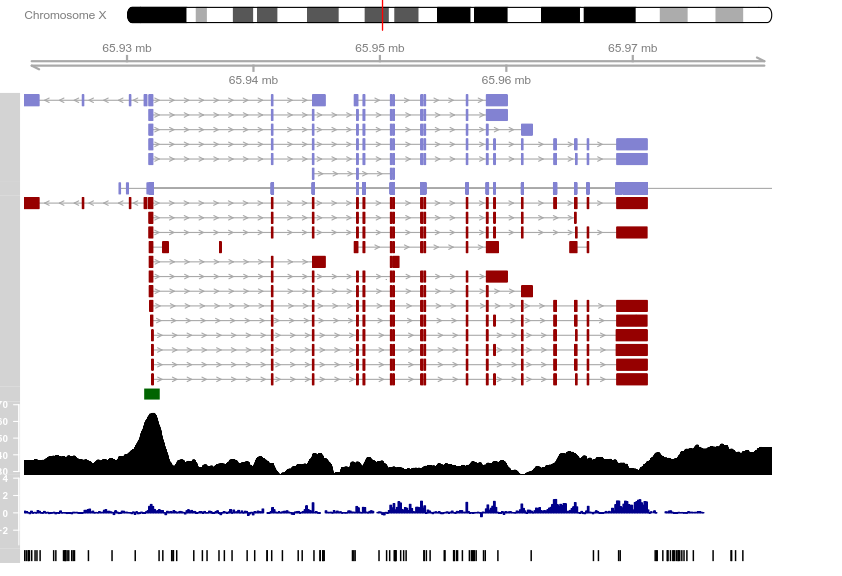
from <- 65921878
to <- 65980988
knownGenes <- UcscTrack(genome = "mm9", chromosome = "chrX",
track = "knownGene", from = from, to = to, trackType = "GeneRegionTrack",
rstarts = "exonStarts", rends = "exonEnds", gene = "name",
symbol = "name", transcript = "name", strand = "strand",
fill = "#8282d2", name = "UCSC Genes")
refGenes <- UcscTrack(genome = "mm9", chromosome = "chrX",
track = "xenoRefGene", from = from, to = to,
trackType = "GeneRegionTrack", rstarts = "exonStarts",
rends = "exonEnds", gene = "name", symbol = "name2",
transcript = "name", strand = "strand", fill = "#8282d2",
stacking = "dense", name = "Other RefSeq")
ensGenes <- UcscTrack(genome = "mm9", chromosome = "chrX",
track = "ensGene", from = from, to = to, trackType = "GeneRegionTrack",
rstarts = "exonStarts", rends = "exonEnds", gene = "name",
symbol = "name2", transcript = "name", strand = "strand",
fill = "#960000", name = "Ensembl Genes")
cpgIslands <- UcscTrack(genome = "mm9", chromosome = "chrX",
track = "cpgIslandExt", from = from, to = to,
trackType = "AnnotationTrack", start = "chromStart",
end = "chromEnd", id = "name", shape = "box",
fill = "#006400", name = "CpG Islands")
snpLocations <- UcscTrack(genome = "mm9", chromosome = "chrX",
track = "snp128", from = from, to = to, trackType = "AnnotationTrack",
start = "chromStart", end = "chromEnd", id = "name",
feature = "func", strand = "strand", shape = "box",
stacking = "dense", fill = "black", name = "SNPs")
conservation <- UcscTrack(genome = "mm9", chromosome = "chrX",
track = "Conservation", table = "phyloP30wayPlacental",
from = from, to = to, trackType = "DataTrack",
start = "start", end = "end", data = "score",
type = "hist", window = "auto", col.histogram = "darkblue",
fill.histogram = "darkblue", ylim = c(-3.7, 4),
name = "Conservation")
gcContent <- UcscTrack(genome = "mm9", chromosome = "chrX",
track = "GC Percent", table = "gc5Base", from = from,
to = to, trackType = "DataTrack", start = "start",
end = "end", data = "score", type = "hist", window = -1,
windowSize = 1500, fill.histogram = "black",
col.histogram = "black", ylim = c(30, 70), name = "GC Percent")
axTrack <- GenomeAxisTrack()
idxTrack <- IdeogramTrack(genome = "mm9", chromosome = "chrX")
plotTracks(list(idxTrack, axTrack, knownGenes, refGenes,
ensGenes, cpgIslands, gcContent, conservation,
snpLocations), from = from, to = to, showTitle = FALSE)