引用自https://mp.weixin.qq.com/s?__biz=MzU4NjU4ODQ2MQ==&mid=2247484662&idx=1&sn=194668553f954e231f4526f5c944a195&chksm=fdf84cb4ca8fc5a2c0e8355377f9d6abdc4fa36b304aa8c533b5e82e49de30d443366ff3346a&mpshare=1&scene=1&srcid=09097IKbscaRGGY4s7Sxju52#rd
Bioconductor应用领域之基因芯片
Bioconductor的起源就是基因芯片分析,因此提供了芯片数据库如GEO、ArrayExpress结口,方便获取数据
芯片提供商:Affymetrix、Illumina、Nimblegen、Agilent
-
Affemetrix 3‘-biased Arrays芯片数据需要affy、gcrma、affyPLM包,这三个包又依赖于芯片定义(CDF)、探针(Probe)、注释(Annotation)三个包,后三个会随前三个安装而自动装好;另外还需要手动安装ROOT包,这个包可以直接使用Affymetrix的数据文件,支持的格式有CDF、PGF、CLF、CSV
-
Affymetrix Exon ST Arrays与Affymetrix Gene ST Arrays芯片需要oligo包和xps包。Ologo包又需要调用pdInfoBuilder包来创建一个pdInfoPackage包,pdInfoPackage包的作用就是合并CDF、Probe、Annotation三种数据
-
Affymetrix SNP Arrays、Affymetrix Tilling Arrays、Nimblegen Arrays芯片数据处理只需要oligo包
-
Ilumina Expression Microarrays芯片数据需要lumi和beadarray包,这两个包都有各自的映射包和注释包:lumi需要lumiHumanAll.db、lumiHumanIDMapping包;beadarray需要illuminaHumanv1BeadID.db、illuminaHumanv1.db包
芯片类型:基因表达芯片、外显子芯片、拷贝数变异检测芯片、SNP芯片、DNA甲基化芯片等
芯片分析内容:预处理、质量评估、差异基因表达分析、基因集富集分析、遗传基因组学
Bioconductor应用领域之测序数据
R也可以处理多种类型数据:fasta、fastq、SAM、BAM、Gff、Bed、Wig等,可以进行测序结果预处理(去除低质量、序列污染等)、格式转换、序列比对、测序质量评估、RNA-seq、差异表达分析、ChIP-seq等
-
SRAdb包是SRA数据库的接口
-
ShortRead包读入测序数据,进行质控、预处理;Rsamtools将数据比对到参考基因组/转录组,完成格式转变和比对结果统计;rtracklayer包将数据导入USCS基因组浏览器(http://genome-asia.ucsc.edu/)进行浏览、操作、输出
-
IRanges、GenomicRanges、genomeIntervals包是基于DNA区域(如染色体)进行数据操作的拓展包;Biostrings包含了序列比对、模式匹配等基本序列分析函数;BSgenome针对有注释的全基因组数据进行操作;GenomicFeatures是对基因组中序列特征(如非编码区)进行注释
-
RNA-seq可用的包有:limma(金标准)、edgeR、DESeq、DEXSeq(外显子水平)、baySeq
-
ChIP-seq:基序发现(Motif discovery)、结合位点检测(Peak calling),可以用CSAR、chipseq、ChIPseqR、ChIPsim、ChIPpeakAnno、DiffBind、iSeq、rGADEM、segmentSeq、BayesPeak、PICS
更多的数据处理,可以看https://www.bioconductor.org/packages/release/BiocViews.html=》softeware=〉technology
Bioconductor应用领域之注释
注释既需要注释工具,有需要链接注释数据库的接口,大体的注释方式有三类
第一类:基于AnnotationDbi包的扩展包
绝大部分注释包都依赖AnnotationDbi包,一旦biocLite安装任何一个“.db”的注释包,AnnotationDbi包都会自动安装。这个包可以创建、操作“.db”,并且可以创建个性化的芯片平台。它又可以分为三类注释包
-
物种型注释包:org.Mm.eg.db、org.Hs.eg.db、org.Rn.eg.db等
包括整个物种相关的基因数据,命名格式:"org.Xx.yy.db", Xx是种属的缩写(注意首字母大写),“yy“来源数据库ID,这个ID用于把所有数据连在一起。如:“org.Hs.eg.db”中Hs是种属,eg是数据库ID -
平台型注释包:如Affymetrix公司的hgu133plus2.db
依赖于具体的实验平台,命名格式:“platformName.db”。“platform”是芯片平台名称,如“hgu95av2.db”就是利用了Affymetrix公司的hgu95av2基因芯片;另外每个“.db”包还需要对应的“.cdf”、“.probe”,名称分别为:“platformName.cdf”、“platformName.probe” -
系统生物学注释包:用于下游分析,如基因功能分析(与上游),比如KEGG.db用来获取KEGG数据库的数据;GO.db用来获取GO数据库的数据;PFAM.db用来获取不同蛋白家族的特征和相关性
第二类:基于biomaRt包获取注释
它与AnnotationDbi包虽然注释信息都是来自远程数据库,但是AnnotationDbi是将注释包本地化,而biomaRt只提供了一组数据接口,加载注释内容严重依赖网络,并且无需维护,不占用本地存储,注释信息自动更新。但它的功能最为强大,因为可以连接各个主要的是给你想你数据库来获取注释信息和数据
Bioconductor应用领域之高通量实验
包括流式细胞仪(Flow Cytometry)、定量PCR、质谱(Mass soectrometry)、蛋白质组及其他基于细胞水平的高通量实验数据
例如定量PCR:HTqPCR、ddCt、qpcrNorm提供如何分析循环阈值(Cycle threshold)的方法
. 利用Bioconductor分析芯片数据
芯片分析主要就是利用bioconductor,分析的过程也体现了它的设计理念和编程思想
基因芯片背景知识
以Affymetrix为例,一个芯片可以包括上百万探针(通常由25个碱基组成),被整齐印刷在芯片上。
-
探针组:一个探针组通常由20对个或11个探针对组成,来自一个基因
-
探针对:匹配探针(Perfect match, PM)+错配探针(Mismatch, MM)。二者仅仅是中间的那个碱基不同。并非所有芯片都有这两个探针,比如外显子芯片,每个探针组只有4个PM探针,没有MM探针
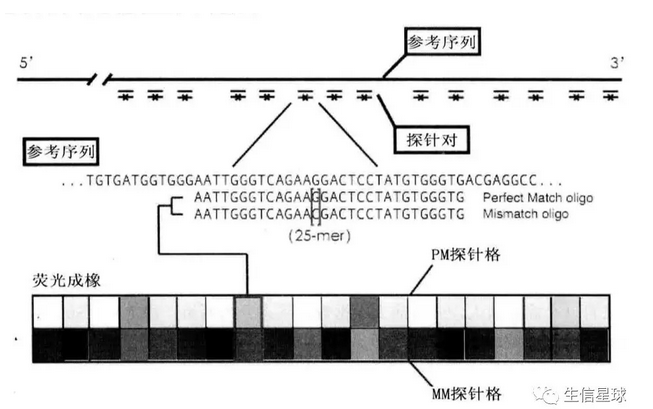
- 探针与探针对
-
探针序列来源:公共核酸数据库中的参考序列(如NCBI的GenBank或RefSeq)
-
不同的芯片中,探针组在参考序列中的分布不同,也就是检测范围不同。3‘表达谱芯片探针组排列在参考序列3’末端附近的一至两个外显子上;外显子芯片中,每个长度大于25个碱基的外显子都有对应的探针组;铺瓦芯片(Tilling array)中,探针基本覆盖了所有的外显子和内含子
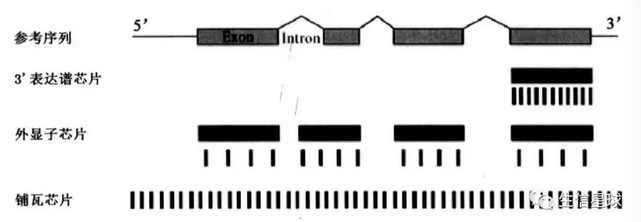
- 探针组与基因
-
探针组与基因关系:芯片数据得到的表达矩阵,实际上是以探针组为单位,而不是直接以基因为单位,每一行对应一个探针组的表达量。后期的分析都是先得到探针组的结果,然后根据注释的ID映射才对应到基因。一般是一个基因同时对应多个探针组。通常会把同一个基因对应的探针组表达量求均值,然后找最大的那个探针组作为代表,让它与该基因一一对应
-
芯片数据获取:一是扫描设备对芯片进行扫描,得到荧光信号图像文件(DAT文件);二是由系统自带的图像处理软件,经过网格定位(Griding)、杂交点范围确定(Spot identifying)、背景噪音过滤(Noise filtering),从芯片图像中提取数据,得到CEL文件
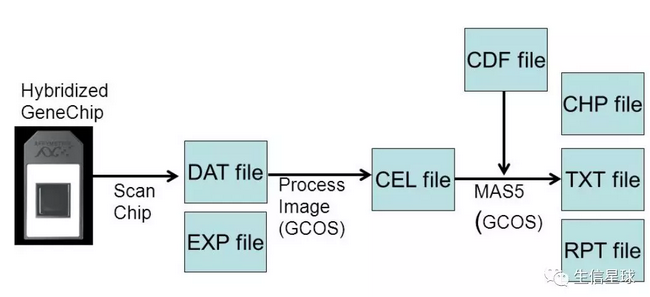
芯片产生的数据文件
关于Affymetrix的数据处理过程:https://slideplayer.com/slide/4804237/
-
CEL文件:CEL文件是Affymetrix最常用的格式,它的主要内容就是每个“cell”的灰度信息,而“cell”就是整个芯片图像划分后得到的小网格,每个小网格中的图像被看作来自一个探针。【补充】当然,如果要得到芯片上每个探针组对应的表达数据,还需要探针的排布信息(哪个探针对应哪个探针组),这部分信息就存储在CDF文件中。要想知道探针对应的序列信息,就需要用Probe文件

数据文件
完成一个小任务
从CLL数据包载入芯片数据=》预处理(先进行一个背景校正)=〉获得探针组表达矩阵
先说一下这个CLL数据集,它包含的是慢性淋巴细胞白血病(Chromic lymphocytic leukemia, CLL)的数据,采用Affymetrix的hgu95av2表达谱芯片(含有12625个探针组),共24个样品("CLL1.CEL"-"CLL24.CEL"),每个样品来自不同的癌症病人。
根据健康状态分为两组(对照试验):稳定组(stable)和恶化组(progressive),每组各12个(平行试验)。
得到的e矩阵是一个12625行、24列的探针组表达矩阵
#安装并加载
source("https://bioconductor.org/biocLite.R")
options(BioC_mirror="http://mirrors.ustc.edu.cn/bioc/")
biocLite("CLL")
library(CLL)
#读入示例数据
data("CLLbatch")
# rma方法进行背景校正【当MM值比PM值还要高时,MM就是杂信号,也就是背景噪声,需要去除】
CLLrma <- rma(CLLbatch)
e_before <- exprs(CLLbatch)
e_after <- exprs(CLLrma)
#对比一下校正前后数据
e_before[1:5,1:5]
e_after[1:5,1:5]