1、安装bioconductor及go分析涉及的相关包
source("http://bioconductor.org/biocLite.R")
options(BioC_mirror="http://mirrors.ustc.edu.cn/bioc/")
biocLite("DO.db", type = "source")
biocLite("BiocUpgrade")
biocLite('clusterProfiler')
biocLite("fastmatch", type = "source")
biocLite("gridExtra")
biocLite("ggraph")
biocLite("UpSetR")
biocLite("cowplot")
biocLite("ggridges")
biocLite("org.Hs.eg.db")
biocLite("topGO")
biocLite("Rgraphviz")
在这一步经常出现以下报错:
报错类型1:Warning message: In file(filename, "r", encoding = encoding) : InternetOpenUrl failed: '操作超时'。
这类报错的解决方法就是修改镜像。R修改镜像步骤:
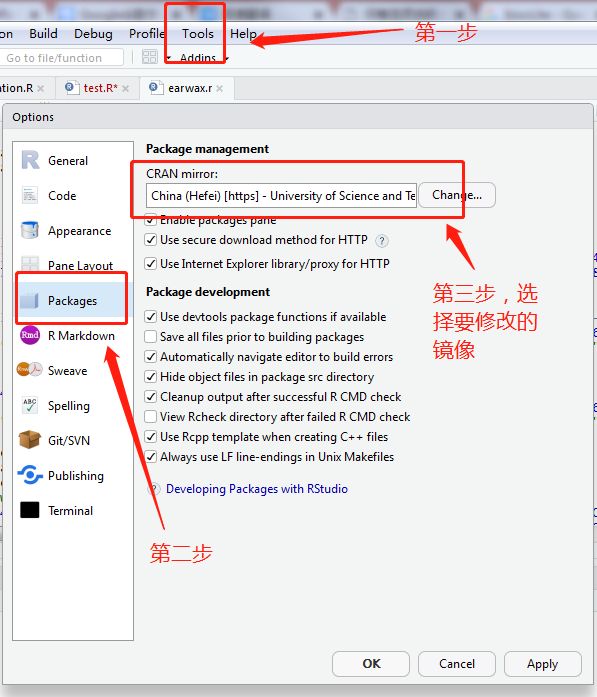
打开RStudio,依次点击:Tools → Packages → CRAN mirror
换镜像的原则是,哪个镜像方便安装哪个包就用哪个镜像
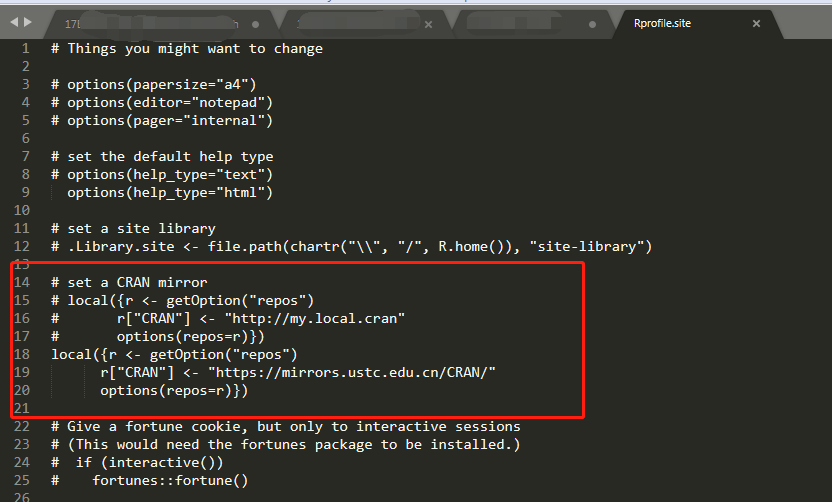
还有第二种修改镜像的方法就是查看R安装在哪儿,修改Rprofile.site.
R.home()
根据上述命令指出来的路径修改镜像
查找X:XXXR-X.X.Xetc 路径下的Rprofile.site文件,用记事本的方式打开,写入如下命令:
# set a CRAN mirror
local({r <- getOption("repos")
r["CRAN"] <- "https://mirrors.ustc.edu.cn/CRAN/"
options(repos=r)})
报错类型2:package ‘XXX’ is not available (for R version X.X.X)
解决这类报错常见思路:1、查看安装包名字有没有写错;2、版本是太新还是太旧,一般而言,都是版本太旧导致的;3、这个安装包是不是一个依赖包,比如"cowplot"是一个依赖于bioconductor的包,就不能直接用“install.packages("cowplot")”这种方式安装,要先安装好bioconductor,再安装biocLite("cowplot");
2、go基因富集分析
如果你的原始数据为ENTREZID格式,则不需要通过bitr转化,如果不是的话,需要通过bitr转化为ENTREZID格式。如果你不知道啥是ENTREZID和SYMBOL,见下面的例子。
ENTREZID SYMBOL Gene name location
1501 CTNND2 catenin delta 2 5p15.2
154664 ABCA13 ATP binding cassette subfamily A member 13 7p12.3
1585 CYP11B2 cytochrome P450 family 11 subfamily B member 2 8q24.3
go基因富集分析正式开始:
install.packages(xlsx)
install.packages(readxl)
library(xlsx)
library(readxl)
model<-read.xlsx("F:/download/38gene.xlsx",1,header=F)
model<-data.frame(model)
library(clusterProfiler)
GENEID_model = bitr(model, fromType="SYMBOL", toType="ENTREZID", OrgDb="org.Hs.eg.db")
#GENID_LIST=as.vector(t(GENEID_model))
go_model <- enrichGO(GENEID_model, OrgDb = org.Hs.eg.db, ont='ALL',pAdjustMethod = 'BH',pvalueCutoff = 0.05,
qvalueCutoff = 0.2,keyType = 'ENTREZID')
write.csv((as.data.frame(go_model)),"F:/download/GENEID_model.csv",row.names =F)
###go富集结果barplot图
barplot(go_model,showCategory=20,drop=T)
####go富集结果点图
dotplot(go_model,showCategory=50)
###绘制GO的网络关系图
go.BP <- enrichGO(go_model, OrgDb = org.Hs.eg.db, ont='CC',pAdjustMethod = 'BH', pvalueCutoff = 0.05, qvalueCutoff = 0.2, keyType = 'ENTREZID')
plotGOgraph(go.BP)
###ont='CC'也可以改为ont='BP'或ont='MF'