PrediXcan , SPrediXcan,MetaXcan是近些年基于GWAS后续分析开发出来的工具。
主要功能是在组织和表达的层面预测影响表型的基因,弥补了GWAS只能在基因组层面解释表型的不足。
下面是这几个工具的工作流程:

今天给大家介绍一下如何使用SPrediXcan和MetaXcan工具进行全转录组分析(Transcriptome-Wide Analysis)
该工具最大的优点是不需要个体水平的基因型数据和表型数据,只需要提供GWAS的summary统计文件就可以预测影响表型的靶基因。
缺点是对电脑的配置要求比较高。
1、配置MetaXcan环境
下载、解压MetaXcan:
wget https://github.com/hakyimlab/MetaXcan/archive/master.zip
unzip master.zip
cd MetaXcan-master/software/
使用conda配置MetaXcan的环境(没安装conda的请自行安装后再做这一步):
conda env create -n MetaXcan -f conda_env.yaml
激活MetaXcan
conda activate MetaXcan
2、配置gwasimputation环境
下载、解压gwasimputation:
wget https://github.com/hakyimlab/summary-gwas-imputation/archive/master.zip
unzip master.zip
配置环境:
conda env create -n gwasimputation -f conda_env.yaml
conda activate gwasimputation
conda install -c conda-forge pyliftover=0.4
conda install -c conda-forge pandas=0.25.3
conda install -c conda-forge scipy=1.4.1
conda install -c conda-forge numpy=1.18.1
conda install -c conda-forge bgen_reader=3.0.2
conda install -c conda-forge cyvcf2=0.20.0
conda install -c conda-forge pyarrow=0.9.0
各位仔细观察的话,会发现我指定了安装包的版本。
建议各位严格安装这个教程的版本安装,不然后面会出现各种因版本过高或过低的报错。
3、下载、解压测试数据
nohup wget https://zenodo.org/record/3657902/files/sample_data.tar?download=1 &
tar -xvpf sample_data.tar?download=1
解压以后,会出现如下的数据:
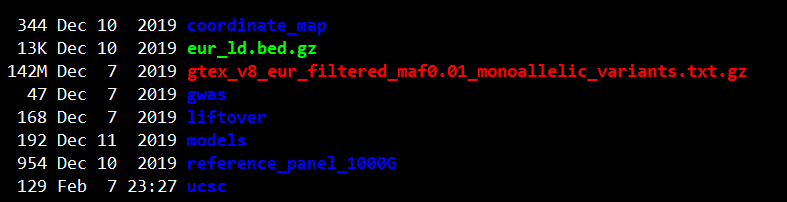
gwas文件夹包含GWAS结果文件的测试数据cad.add.160614.website.txt.gz;
完成以上工作以后,我们就可以开始分析数据啦!
以下是全转录组分析的流程
4、协调GWAS文件的坐标(hg19坐标统一为hg38)
原始GWAS结果文件cad.add.160614.website.txt.gz的格式如下所示:
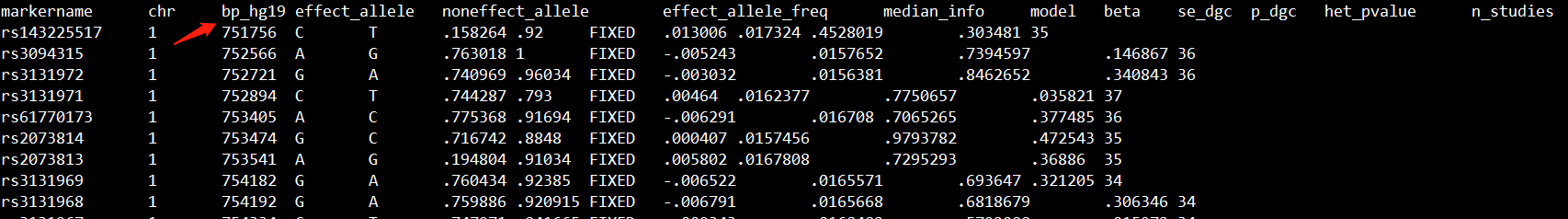
我们可以看到,这个文件的坐标是hg19版本的,但MetaXcan的最新版是hg38,因此我们需要先将GWAS结果文件cad.add.160614.website.txt.gz的坐标转为hg38。
这里面,我们需要先定义几个文件夹:
GWAS_TOOLS=/chenwenyan/software/gwas-imputation-master/src #步骤二的gwas-imputation-master的路径
DATA=/chenwenyan/anaconda2/envs/gwasimputation/data/data #步骤三下载的测试数据的路径
OUTPUT=/chenwenyan/test #这个自己定义:输出文件的路径
注意,以上路径用的是我自己的路径(比如
/chenwenyan/software/gwas-imputation-master/src),不要照搬。请修改成你自己的路径。
定义完路径以后,在输出文件夹$OUTPUT(我的示例是/chenwenyan/test)下新建一个文件夹创建文件夹harmonized_gwas,输入命令:mkdir harmonized_gwas
随后,创建一个test.sh的文件,输入命令:
vi test.sh
在test.sh文件输入以下内容:
#!/bin/bash
GWAS_TOOLS=/chenwenyan/software/gwas-imputation-master/src #gwas-imputation-master的路径
DATA=/chenwenyan/anaconda2/envs/gwasimputation/data/data #下载的测试数据的路径
OUTPUT=/chenwenyan/test #输出文件的路径
python $GWAS_TOOLS/gwas_parsing.py
-gwas_file $DATA/gwas/cad.add.160614.website.txt.gz
-liftover $DATA/liftover/hg19ToHg38.over.chain.gz
-snp_reference_metadata $DATA/reference_panel_1000G/variant_metadata.txt.gz METADATA
-output_column_map markername variant_id
-output_column_map noneffect_allele non_effect_allele
-output_column_map effect_allele effect_allele
-output_column_map beta effect_size
-output_column_map p_dgc pvalue
-output_column_map chr chromosome
--chromosome_format
-output_column_map bp_hg19 position
-output_column_map effect_allele_freq frequency
--insert_value sample_size 184305 --insert_value n_cases 60801
-output_order variant_id panel_variant_id chromosome position effect_allele non_effect_allele frequency pvalue zscore effect_size standard_error sample_size n_cases
-output $OUTPUT/harmonized_gwas/CARDIoGRAM_C4D_CAD_ADDITIVE.txt.gz
GWAS结果文件的染色体用数值表示,不要用
chr+数值的方式GWAS文件格式必须是gz压缩格式,分隔符为tab
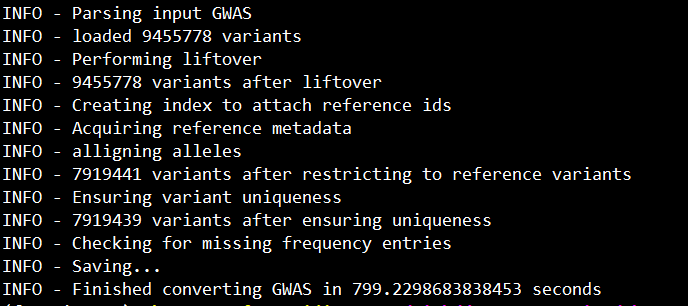
运行:sh test.sh
顺利的话,会输入以下信息:
生成的新文件如下所示:
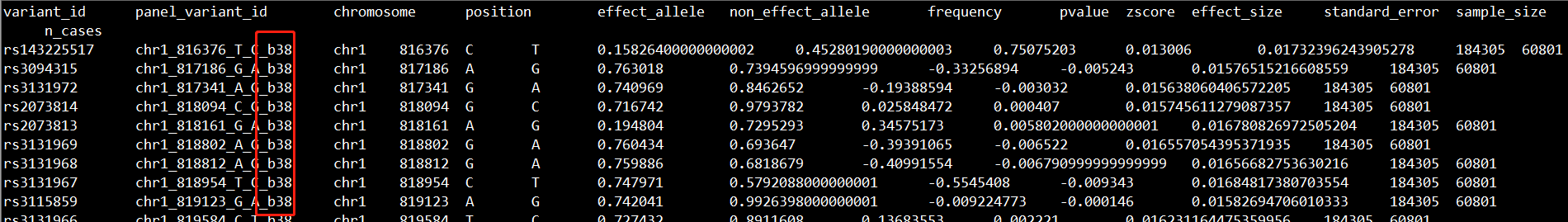
如截图的红框所示,此时坐标已变为hg38了。
5、对gwas结果文件进行imputation
先在输出文件夹$OUTPUT(我的示例是/chenwenyan/test)下新建一个文件夹summary_imputation,输入命令:mkdir summary_imputation
随后输入如下命令进行imputation:
GWAS_TOOLS=/chenwenyan/software/gwas-imputation-master/src
DATA=/chenwenyan/anaconda2/envs/gwasimputation/data/data
OUTPUT=/chenwenyan/test
python $GWAS_TOOLS/gwas_summary_imputation.py
-by_region_file $DATA/eur_ld.bed.gz
-gwas_file $OUTPUT/harmonized_gwas/CARDIoGRAM_C4D_CAD_ADDITIVE.txt.gz
-parquet_genotype $DATA/reference_panel_1000G/chr1.variants.parquet
-parquet_genotype_metadata $DATA/reference_panel_1000G/variant_metadata.parquet
-window 100000
-parsimony 7
-chromosome 1
-regularization 0.1
-frequency_filter 0.01
-sub_batches 10
-sub_batch 0
--standardise_dosages
-output $OUTPUT/summary_imputation/CARDIoGRAM_C4D_CAD_ADDITIVE_chr1_sb0_reg0.1_ff0.01_by_region.txt.gz
出现如下页面说明顺利运行了:
上面示例只是对一号染色体上的第一个批次进行imputation,我们实际上要进行220次(22条染色体*10个批次)循环imputation。
因此跑完上面流程以后,需要继续跑剩下的119次imputation。
需要修改的地方有
$DATA/reference_panel_1000G/chr1.variants.parquet、-chromosome 1、-sub_batch 0、$OUTPUT/summary_imputation/CARDIoGRAM_C4D_CAD_ADDITIVE_chr1_sb0_reg0.1_ff0.01_by_region.txt.gz
$DATA/reference_panel_1000G/chr1.variants.parquet修改的数值是chr1,chr2,chr3,……,chr22;
-chromosome 1修改的数值是:1,2,3,……,22;
-sub_batch 0修改的数值是:0,1,2,……,9;
$OUTPUT/summary_imputation/CARDIoGRAM_C4D_CAD_ADDITIVE_chr1_sb0_reg0.1_ff0.01_by_region.txt.gz是输出文件,建议修改chr1_sb0为对应的染色体和批次。
举个例子,我现在想跑3号染色体的第十个批次,那么脚本应该如下所示:
GWAS_TOOLS=/chenwenyan/software/gwas-imputation-master/src
DATA=/chenwenyan/anaconda2/envs/gwasimputation/data/data
OUTPUT=/chenwenyan/test
python $GWAS_TOOLS/gwas_summary_imputation.py
-by_region_file $DATA/eur_ld.bed.gz
-gwas_file $OUTPUT/harmonized_gwas/CARDIoGRAM_C4D_CAD_ADDITIVE.txt.gz
-parquet_genotype $DATA/reference_panel_1000G/chr3.variants.parquet
-parquet_genotype_metadata $DATA/reference_panel_1000G/variant_metadata.parquet
-window 100000
-parsimony 7
-chromosome 3
-regularization 0.1
-frequency_filter 0.01
-sub_batches 10
-sub_batch 9
--standardise_dosages
-output $OUTPUT/summary_imputation/CARDIoGRAM_C4D_CAD_ADDITIVE_chr3_sb9_reg0.1_ff0.01_by_region.txt.gz
6、合并220个批次的数据
输入如下命令:
GWAS_TOOLS=/chenwenyan/software/gwas-imputation-master/src
DATA=/chenwenyan/anaconda2/envs/gwasimputation/data/data
OUTPUT=/chenwenyan/test
python $GWAS_TOOLS/gwas_summary_imputation_postprocess.py
-gwas_file $OUTPUT/harmonized_gwas/CARDIoGRAM_C4D_CAD_ADDITIVE.txt.gz
-folder $OUTPUT/summary_imputation
-pattern CARDIoGRAM_C4D_CAD_ADDITIVE.*
-parsimony 7
-output $OUTPUT/processed_summary_imputation/imputed_CARDIoGRAM_C4D_CAD_ADDITIVE.txt.gz
和上面一样,路径不要照搬我的,请修改成你自己的路径。
正常运行的话,会出现如下界面:

7、使用单个组织预测基因表达
在输出文件夹$OUTPUT(我的示例是/chenwenyan/test)下新建一个文件夹spredixcan,输入命令:mkdir spredixcan
在spredixcan文件夹下新建文件夹eqtl,输入命令:mkdir eqtl
建完后,输入如下命令:
DATA=/chenwenyan/anaconda2/envs/gwasimputation/data/data
METAXCAN=/chenwenyan/software/MetaXcan-master/software
python $METAXCAN/SPrediXcan.py
--gwas_file $OUTPUT/processed_summary_imputation/imputed_CARDIoGRAM_C4D_CAD_ADDITIVE.txt.gz
--snp_column panel_variant_id --effect_allele_column effect_allele --non_effect_allele_column non_effect_allele --zscore_column zscore
--model_db_path $DATA/models/eqtl/mashr/mashr_Whole_Blood.db
--covariance $DATA/models/eqtl/mashr/mashr_Whole_Blood.txt.gz
--keep_non_rsid --additional_output --model_db_snp_key varID
--throw
--output_file $OUTPUT/spredixcan/eqtl/CARDIoGRAM_C4D_CAD_ADDITIVE__PM__Whole_Blood.csv
正常运行的话,会出现如下界面:

8、使用多个组织预测基因表达
在输出文件夹$OUTPUT(我的示例是/chenwenyan/test)下新建一个文件夹smultixcan,输入命令:mkdir smultixcan
在smultixcan文件夹下新建文件夹eqtl,输入命令:mkdir eqtl
python $METAXCAN/SMulTiXcan.py
--models_folder $DATA/models/eqtl/mashr
--models_name_pattern "mashr_(.*).db"
--snp_covariance $DATA/models/gtex_v8_expression_mashr_snp_covariance.txt.gz
--metaxcan_folder $OUTPUT/spredixcan/eqtl/
--metaxcan_filter "CARDIoGRAM_C4D_CAD_ADDITIVE__PM__(.*).csv"
--metaxcan_file_name_parse_pattern "(.*)__PM__(.*).csv"
--gwas_file $OUTPUT/processed_summary_imputation/imputed_CARDIoGRAM_C4D_CAD_ADDITIVE.txt.gz
--snp_column panel_variant_id --effect_allele_column effect_allele --non_effect_allele_column non_effect_allele --zscore_column zscore --keep_non_rsid --model_db_snp_key varID
--cutoff_condition_number 30
--verbosity 7
--throw
--output $OUTPUT/smultixcan/eqtl/CARDIoGRAM_C4D_CAD_ADDITIVE_smultixcan.txt
9、结果演示
生成的结果如下所示:

结果文件标题所代表的意思:
gene: Ensembl id;
gene_name: HUGO gene name;
Z-score: S-PrediXcan's association result for the gene;
pvalue: P-value of the aforementioned statistic;
var_g: variance of the gene expression, calculated as W' * G * W (where W is the vector of SNP weights in a gene's model, W' is its transpose, and G is the covariance matrix);
pred_perf_r2: R2 of tissue model's correlation to gene's measured transcriptome (prediction performance); pred_perf_pval: pval of tissue model's correlation to gene's measured transcriptome (prediction performance);
n_snps_used: number of snps from GWAS that got used in S-PrediXcan analysis;
n_snps_in_cov: number of snps in the covariance matrix;
n_snps_in_model: number of snps in the model.
结果文件的pvalue值超过Bonferroni correction的阈值就说明该结果有意义。
Bonferroni correction定义:0.05/(N个基因*M个组织)
