SNP的rsid匹配
在处理 Nealelab 中的summary data sets时,发现数据缺失SNP对应rs号:
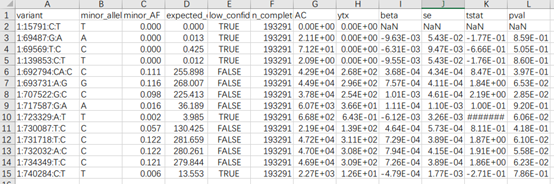
可以看到数据中只有variant变量,这里提供了解决方案:https://www.biostars.org/p/349284/ ,实践一下!
Getting Started
首先下载对应的SNP注释信息
Common SNPs
VCFcommon=ftp://ftp.ncbi.nih.gov/snp/organisms/human_9606_b151_GRCh38p7/VCF/common_all_20180418.vcf.gz
wget -b -c ${VCFcommon}
All SNPs:
VCFall=ftp://ftp.ncbi.nih.gov/snp/organisms/human_9606_b151_GRCh38p7/VCF/All_20180418.vcf.gz
wget -b -c ${VCFall}
在R中读取数据
下载了全部的数据以防万一,VCF数据读取(很慢。。。)
读取数据很慢,读取中是这样显示的:
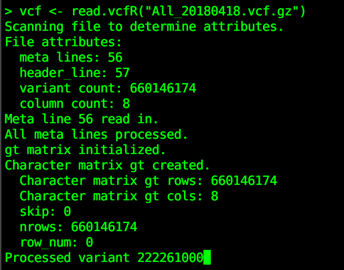
15G读不出来,换成common SNPs(1.5G):
成功了,导出txt格式方便使用
install.packages(“vcfR”)
library(vcfR)
vcf <- read.vcfR("common_20180418.vcf.gz")
vcf2 = vcf@fix[,1:3]
fwrite(vcf2,"common_all_20180418.txt",sep = " ",quote = T,row.names = F,col.names = T)
Day 2
今天匹配的时候出问题了,全都匹配不上,查询后发现可能是版本号不一致
数据中的RSid用的是hg19 (GRCh37) 而我下载的却是hg38 (GRCh38),真坑,重新下载
Common SNPs
VCFcommon=ftp://ftp.ncbi.nih.gov/snp/organisms/human_9606_b151_GRCh37p13/VCF/common_all_20180423.vcf.gz
wget -b -c ${VCFcommon}
可以,成功与summary data匹配上!
在这发现一个新的方法,回头再试一下:https://www.cnblogs.com/chenwenyan/p/13166529.html