老生常谈的scRNASeq基本操作和原理:

scrnaseq 的测序方法发展的年表:
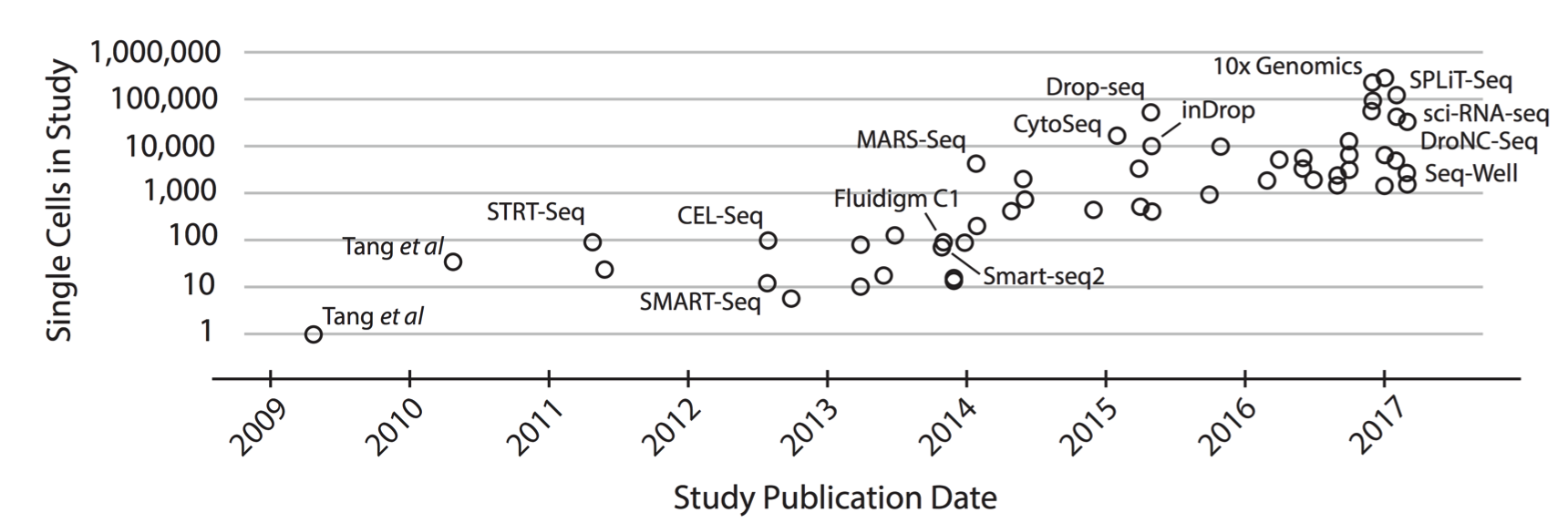
在看一下列表:

分选单细胞的96孔板:

分析前的常规数据处理:FastQC、Trimming Reads、Demultiplexing(identify cell containing droplets)、using STAR to align(with genome)、 Kallisto align(align without genome,using k-mers method)、construct expression matrix(with UMIs)、Bioconductor to analysis the raw data(SingleCellExperiment,scater,ggplot2)、Expression QC、data visualization、identify confounding factors、normalized expression data
得到均一化之后的matrix之后:
Biological analysis:Clustering 、Feature selection、Pseudotime analysis、Imputation、DE
Seurat:Create seurat object、Normalization、Highly variable genes、dealing with confounders、linear dimensionality reduction、significant PCs、clustering cells、markers genes
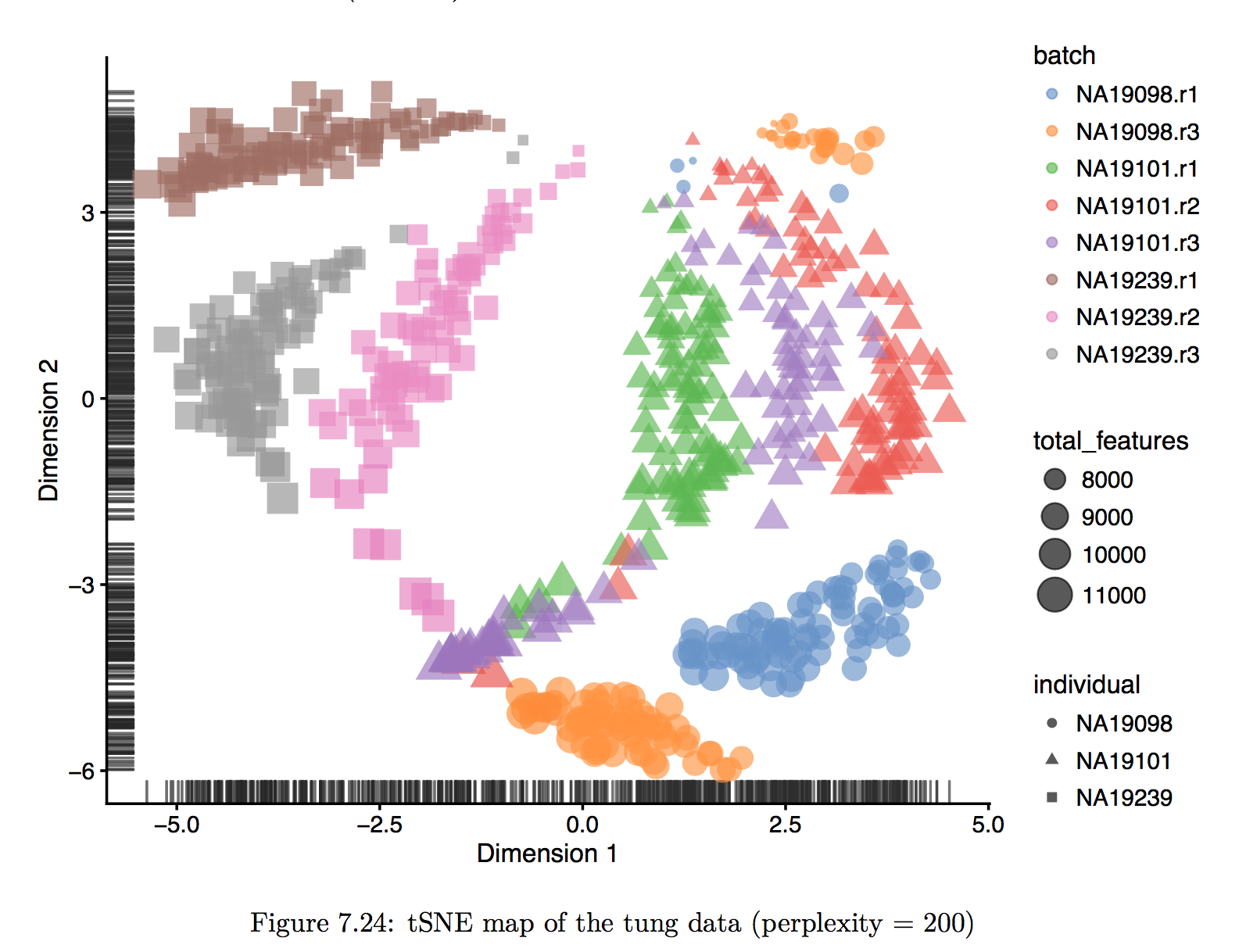


主要讲一讲降维分析中的Seurat:
需要在R里面加载的包:

用小提琴图以及基因图来做一些数据的观测之后(Vlnplot、Geneplot、FilterCells),对数据做normalized(NormalizeData)、以及find highly variable genes(FindVariableGenes)、dealing with confounders(ScaleData)、LDR(RunPCA、printPCA、PCAPlot、PCHeatmap)、Significant PCs(JackStraw、JackStrawPlot、PCEIbowPlot)、clustering cells(FIndClusters、PrintFindClusterParams、adjustedRandIndex、RunTSNE、TSNEPlot)、marker genes(FIndMarkers、VlnPlot、FeaturePlot、FIndALLMarkers)

两种marker genes的violin plot以及全部marker genes的tSNEplot
整体Scrnaseq date的分析流程和思路是很清晰明确的,本教程几乎满足了所有scrnaseq中目前遇到的问题和解决方法,可以说是一本单细胞下机数据分析以及degub的Brochure