4.NGS中的reads mapping
顾名思义,就是将测序的得到的DNA定位在基因组上。
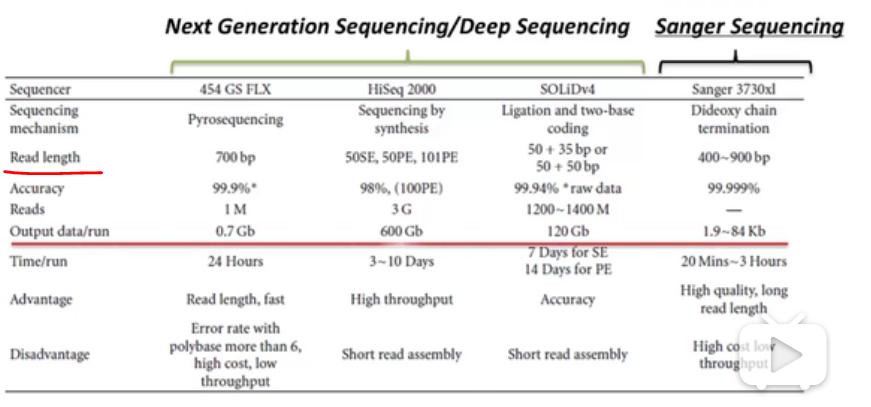
因为二代测序的得到的序列是较短的,reads mapping很好地解决了这个问题。

本质上reads mapping是一个双序列比对问题,但和之前讲的NW和SW的不一样,后者适用于两者长度相差不大的。
现在问题有几个特征:
1.reads和ref的长度有着跨数量级的差异,reads长度通常不超过100bp,而ref基因组通常在上百Mb。
2.数据量,NGS测序产生的数据量达到几百Gb,相当于几十个人的人类基因组。
3.数据质量。在双序列比对中通常假定序列本身不会出错,但是NGS所产生的reads质量参差不齐。
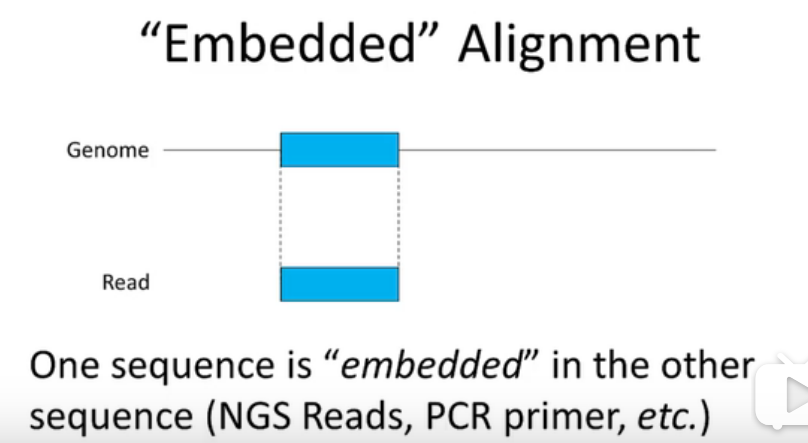
reads可以说是镶嵌到基因组序列中的,对于基因组来说是局部比对,对于reads来说是全局比对,是一个混合型的alignment。
首先对基因组建立索引,也就是index,

将每一个基因根据key映射到一个index,从而存储在不同的数据块中,尽量减少比对时间。
哈希可以来完成,以下例子:

先给ACGT分别确定一个值,那么将求和作为哈希函数,将基因组中分段,然后进行映射存储。这样有一个reads之后就可以以O(1)时间内寻找位置。
通常有一定的容错性。

数据压缩中的前缀树和后缀树被应用于reads mapping。 这里也提到了bowtie和BTW(Burruws Wheeler transform),提高了内存利用效率和比对速度。

在对短序列对比时,将所有的SQ都算出来,read中每个碱基都有一个测序质量,假定错配都是由测序错误引起的,从而计算出SQ。
在实际对read mapping的比对中,通常不使用序列比对分数,而使用mapping Quality(也就是最后一行的E),来筛选Read在Ref中的位置。
//这个415是如何得到的呢?是所有SQ的和。
当将reads正确映射到基因组之后,就可以来判断遗传变异。
根据遗传变异的尺寸,可以分为单个碱基水平的单核苷酸变异和多个:

//这个图说的简直十分清晰。
SNV是最常见的遗传变异分析方法:包括替换碱基,或者插入删除碱基。
SV:包括大规模删除插入、倒转、易位、拷贝数变异。

SNP calling是确定哪个基因位点存在变异,不涉及到对应位点的基因型。
Genotype calling是进一步确定变异位点的基因型是纯合的还是杂合的。
测序深度(sequencing depth):测序得到的碱基总量与基因组大小的比值。 它与基因组覆盖度是一个正相关的关系。测序错误率和假阳性结果会随测序深度的提高而下降。
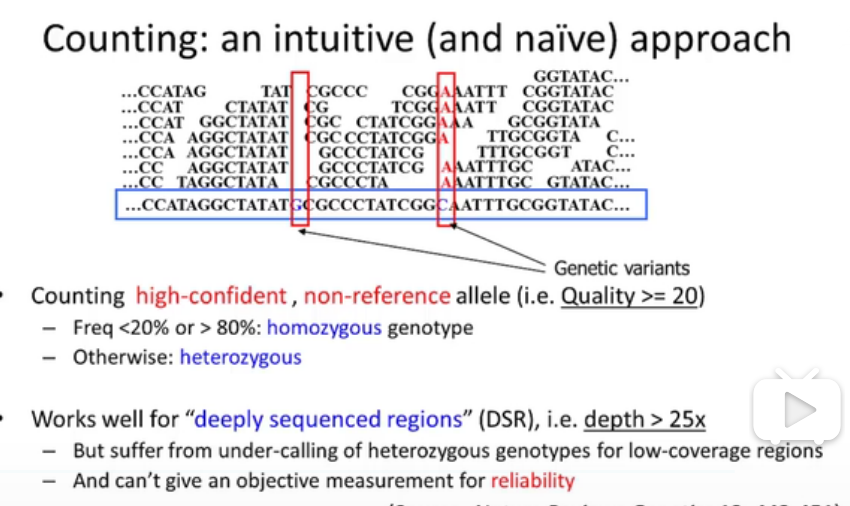
//这张图它在说什么,我完全听不懂啊。什么就是纯和了,怎么就杂合了?

这里给出了一个简单的概率模型。
一个生物体的基因型,有三种情况,那么假设在基因测序中测到的有k个A,有n-k个a。
如果是AA,那么概率就是n-k个a错误概率的乘积,杂合子由1-二者之和。
那么如果知道生物体中三种基因型出现了概率作为先验概率,那么可以推算出,后验概率。
//其实这里不太明白D是什么?